Questi sono appunti della lezione 4 del corso di biologia cellulare di Harvard Extension.
Il percorso secretorio si riferisce al reticolo endoplasmatico, all’apparato di Golgi e alle vescicole che viaggiano in mezzo ad essi, nonché alla membrana cellulare e ai lisosomi. Si chiama “secretoria” perché è la via attraverso la quale la cellula secerne le proteine nell’ambiente extracellulare. Ma come al solito, l’etimologia racconta solo una frazione della storia. Questa via elabora anche le proteine che saranno legate alla membrana (sia nella membrana cellulare che nelle stesse membrane dell’ER o del Golgi), così come gli enzimi lisosomiali, e anche tutte le proteine che vivranno la loro vita nella via secretoria stessa. Fa anche altre cose oltre ad elaborare le proteine.
Il citosol e il ‘lume’ (il liquido che riempie la via secretoria) sono ambienti chimici diversi, e normalmente non si mescolano mai. Il citosol è riduttivo (quando sei nel citosol, continui a incontrare molecole che vogliono offrirti elettroni), e l’ER, il Golgi e l’ambiente extracellulare sono ossidativi (le molecole continuano a venirti incontro chiedendoti elettroni). Vedi redox se sei ancora confuso. Questo fa sì che le condizioni di ripiegamento delle proteine siano diverse: per esempio, i legami disolfuro di solito si formano solo in condizioni ossidative. Inoltre, diverse proteine possono vivere solo nella via secretoria o solo nel citosol. La via secretoria fornisce alla cellula una via per gestire cose che non sarebbe bene avere nel citoplasma, e/o che sono più utili se tenute concentrate in un compartimento specializzato con i loro partner di interazione desiderati. Gli epatociti (nel fegato) sequestrano farmaci e tossine nell’ER liscio e li scompongono per l’escrezione dal corpo. La via secretoria non è contigua, ma ogni movimento tra i suoi componenti avviene in piccoli microcosmi gorgogliati del proprio mondo chimico, chiamati vescicole.
Molte proteine che passano attraverso la via secretoria non toccano mai il citosol – tranne le parti di proteine di membrana che sporgono sul lato citosolico. Molte di loro hanno bisogno di chaperon per aiutare il ripiegamento e/o tutta una serie di modifiche post-traslazionali per essere pronte per la loro funzione nativa, e la via secretoria è specializzata nel fornire loro tutto questo.
La lezione di oggi si concentrerà su come le proteine vengono tradotte nell’ER e come viaggiano (in vescicole) tra l’ER, il Golgi e altre destinazioni. Questo è splendidamente rappresentato nel video Life of the Cell:
Il reticolo endoplasmatico è il primo passo nel percorso secretorio. La sua membrana è continua con la membrana nucleare esterna, anche se non è chiaro perché ciò sia importante, dato che le proteine non iniziano la loro vita nel nucleo. Piuttosto, gli mRNA vanno alla deriva nel citoplasma finché non vengono raccolti da un ribosoma interessato a tradurli. Nella “traslocazione post-traslazionale” la nuova proteina viene spostata nell’ER dopo essere stata tradotta. Nel fenomeno più interessante chiamato “traslocazione cotraslazionale” il ribosoma inizia la traduzione come qualsiasi altra proteina, ma da qualche parte nei primi 16-30 aminoacidi colpisce un peptide di segnale (alias sequenza di segnale). Il motivo del segnale è spesso 1 amminoacido caricato positivamente seguito da 6-12 amminoacidi idrofobici. Questo motivo viene riconosciuto dalla particella di riconoscimento del segnale (SRP, una ‘ribonucleoproteina’ o molecola ibrida RNA/proteina) che si lega ad essa e impedisce al ribosoma di continuare la traduzione. La traduzione si ferma finché il complesso ribosoma/SRP non incontra un recettore SRP sulla membrana ER. Quando si incontrano, SRP e il suo recettore legano ciascuno una molecola di GTP nella membrana ER, che apparentemente rafforza la loro interazione. Fortuitamente, tutto questo accade adiacente a un traslocatore Sec61 – un complesso proteico che forma un canale che attraversa la membrana ER. Il traslocatore è in realtà un complesso di tre diverse proteine (geni: SEC61A1 o SEC61A2, SEC61B, SEC61G), di cui la subunità Sec61a ha 10 a-eliche che si estendono sulla membrana e che formano il canale. Una volta che il ribosoma è agganciato alla membrana, continua la traduzione, spingendo il peptide del segnale ed eventualmente l’intera proteina attraverso il canale nel lume dell’ER. Quando la traduzione si ferma, SRP e il recettore SRP idrolisano entrambi il loro GTP per liberarsi a vicenda e il carico del ribosoma (questo deve richiedere l’energia del GTP, dato che il legame originale era in discesa), una peptidasi del segnale scinde il peptide del segnale dalla proteina nascente, e la proteina è libera di iniziare a piegarsi nell’ER.
Per alcune proteine ER sono coinvolti un paio di altri attori. L’oligosaccaride transferasi, che aggiunge gruppi glicosilici alle asparagine nella proteina nascente, fa parte del complesso translocon e in realtà esegue la glicosilazione mentre la nuova proteina viene ancora tradotta. Quindi, anche se chiamiamo la glicosilazione una “modifica post-traslazionale”, in questo caso viene effettuata durante la traduzione. Inoltre, per ottenere la loro corretta struttura, alcune proteine hanno bisogno di essere completamente tradotte prima che sia permesso loro di iniziare il ripiegamento – se la porzione N-terminale fosse permessa di iniziare il ripiegamento non appena entrata nel lume, finirebbe per avere una struttura generale sbagliata. Per evitare questo, a volte BiP il chaperone lega la proteina per mantenerla spiegata per un po’. Immaginate BiP come un altro Pac-Man che morde la proteina per mantenerla lineare, come Hsc70 nel processo di targeting mitocondriale (vedi settimana scorsa).
Ecco un video di questo:
I primi due minuti mostrano lo scenario di base descritto sopra. Poi si passa a uno scenario più complesso che introdurrò tra un minuto. Per tua informazione, il video mostra due cose “controverse” non incluse nella descrizione di cui sopra: (1) il peptide di segnale che viene degradato nella membrana, e (2) una “proteina spina” che blocca il canale prima/dopo la traduzione. Non tutti gli scienziati sono ancora d’accordo su queste due cose.
Tutte le proteine che sappiamo passano attraverso la via secretoria sono state individuate lì da persone che fanno esperimenti di localizzazione per vedere dove si trova una proteina nella cellula. Un fatto strano dell’ER è che si può mettere la cellula in un frullatore e poi l’ER inizierà a ricollegarsi a se stesso, formando piccoli ‘microsomi’ che non sono attaccati al nucleo ma formano bolle contigue di ER. Si può quindi iniziare a giocare con le proteasi – che scompongono le proteine – e i detergenti – che solubilizzano la membrana ER. Supponendo che la vostra proteina di interesse sia tradotta, potete controllare se (1) sopravvive al trattamento con la proteasi ma (2) non sopravvive al trattamento con proteasi + detergente, allora è una proteina della via secretoria. La logica è che nel caso (1) era protetta all’interno dell’ER, ma nel caso (2) hai sciolto l’ER, quindi è stata mangiata dalla proteasi. Tutto questo presuppone che tu abbia un anticorpo o qualche altro modo di rilevare se la proteina di interesse è lì dopo questi trattamenti.
La gente ha anche usato tali tecniche per capire che solo 70 aminoacidi di una nuova proteina possono essere tradotti prima che diventi troppo tardi per quella proteina per finire nell’ER. Ricordate, il peptide segnale è nei primi 16-30 aminoacidi, e la traslocazione nell’ER dipende dalla presenza di SRP. I ribosomi traducono ad una velocità prevedibile, così la gente ha fatto iniziare i ribosomi a tradurre un po’ di mRNA e poi ha aspettato un certo periodo di tempo prima di aggiungere SRP, per vedere quanta traduzione poteva avvenire prima che SRP non potesse più fare il suo lavoro.
Il recettore SRP e le proteine Sec61 sono proteine di membrana ER – e ci sono anche molte altre proteine di membrana ER, membrana Golgi e lisosoma. Infatti, anche le proteine di membrana (vedi classe 02) della membrana cellulare vengono processate nella via secretoria. Molte di queste hanno diversi o decine di domini transmembrana (20-25 aminoacidi idrofobici ciascuno) che devono essere inseriti nel giusto ordine e orientamento (per esempio, vuoi davvero che i tuoi canali ionici e trasportatori puntino nella giusta direzione, dentro o fuori la cellula). Di conseguenza ci sono un sacco di meccanismi biologici fantasiosi per inserire correttamente queste proteine nella membrana. Questo è ciò che descrive l’ultima metà del video qui sopra.
Ecco una tautologia: alcune proteine hanno una sequenza topogenica che determina il loro orientamento nella membrana. Questa sequenza è composta da due tipi di sequenze di segnale:
- una sequenza di stop-transfer (abbreviata STA per qualche motivo) è una sequenza di 22-25 aminoacidi idrofobici da qualche parte nel mezzo della proteina che forma un’alfa elica. Quando viene incontrata viene spinta nella membrana, e poi la traduzione del resto della proteina continua nel citosol. Quindi questo tipo di “annulla” la traslocazione verso l’ER che è stata avviata dal peptide di segnale all’inizio (terminale N) della proteina.
- una sequenza di ancoraggio del segnale (abbreviato SA) è anche un’alfa-elica idrofoba di 22-25aa, ma con una serie di ~3 amminoacidi caricati positivamente alla sua sinistra o destra. Come il peptide di segnale, questo è riconosciuto da SRP, che porta il ribosoma all’ER. Ma a differenza del peptide di segnale, questa sequenza alfa-elica sarà inserita nella membrana dell’ER. L’orientamento dell’inserimento è determinato dai 3 amminoacidi caricati positivamente. Le cariche positive devono sempre finire sul lato citosolico, quindi se vengono dopo (cioè C-terminale della) sequenza idrofoba, la proteina finisce con la sua estremità terminale C puntata nel citosol, ma se vengono prima (cioè N-terminale della) sequenza idrofoba, la proteina finisce con la sua estremità N puntata nel citosol.
Con questi due segnali come mattoni, si può immaginare una proteina con una serie di sequenze di trasferimento di stop e di ancoraggio del segnale per creare tutta una serie di domini transmembrana avanti e indietro cuciti nella membrana come da una macchina da cucire. Le persone hanno classificato le proteine di membrana in cinque categorie:
- Il tipo I ha solo un peptide di segnale e poi uno stop transfer nel mezzo. Quindi finisce con il suo terminale N (idrofilo) nel lume, il suo centro (idrofobo) nella membrana e il suo terminale C (idrofilo) nel citosol.
- Il tipo II non inizia con un peptide di segnale. Inizia come qualsiasi altra proteina, ma nel mezzo ha una sequenza di ancoraggio del segnale con gli aminoacidi +++ che vengono prima e la serie idrofoba dopo. Questo fa sì che la proteina venga traslata a metà della traduzione, con la parte N-terminale già tradotta che sporge nel citosol (dato che i +++ devono rimanere citosolici) e la parte C-terminale che ora inizia ad essere tradotta viene tradotta direttamente nell’ER. Così finisce transmembrana con il suo terminale C nell’ER e il terminale N nel citosol – opposto al Tipo I.
- Il Tipo III è come il Tipo II – nessun peptide di segnale, solo un’ancora di segnale nel mezzo, ma in questo caso i +++ vengono dopo la sequenza idrofobica, che inverte l’orientamento. Così questo finisce con il suo terminale N nell’ER e il suo terminale C nel citosol. L’opposto del tipo II e, alla fine, lo stesso del tipo I, anche se ci è arrivato in un modo diverso – non ha un peptide di segnale che viene scisso nell’ER.
- Le proteine di tipo IV o ‘multipass’ hanno una serie alternata di sequenze di segnale e sequenze di trasferimento di stop. Queste sono chiaramente più di un ‘tipo’, eppure non sono così diverse come la vostra immaginazione combinatoria potrebbe permettere. L’orientamento della prima sequenza di segnale determina se il terminale N finirà nel citosol o nell’ER, e il numero totale di sequenze di trasferimento di stop + ancora di segnale determina dove finirà il terminale C: un numero pari = stesso lato del terminale N, numero dispari = lato opposto del terminale N. Le sequenze STA e SA devono rigorosamente alternarsi, con l’eccezione che si può iniziare con due sequenze di ancoraggio del segnale se la prima è orientata con il terminale N nel citosol. Giusto per prendere in giro questo schema di categorizzazione, sono stati definiti alcuni sottotipi incompleti di Tipo IV, dove il Tipo IVa è N-terminale nel citosol (quindi inizia come una proteina di Tipo II) e il Tipo IVb è N-terminale nel lume (inizia come una proteina di Tipo III ma poi ha un’altra sequenza SA che la riporta nell’ER). GLUT1 della classe 02 è un Tipo IVa.
- Le proteine ancorate a GPI, che sono il quinto tipo ma non sono chiamate Tipo V, iniziano con un peptide di segnale e terminano con un C-terminale idrofobico che rimane incorporato nella membrana. Quell’estremità idrofoba viene tagliata via e sostituita con GPI, che rimane anch’esso incorporato nella membrana. La PrP è una di queste – ne parleremo più tardi.
A questo punto abbiamo discusso come le proteine possono finire nel lume dell’ER o attraversare la membrana dell’ER. La maggior parte delle proteine lasciano l’ER entro pochi minuti, trasportate in vescicole destinate al Golgi e poi più tardi all’escrezione, ai lisosomi o alla membrana cellulare. Questa direzione di viaggio in avanti è chiamata anterograda; andare indietro dal Golgi all’ER è il trasporto retrogrado.
Entrambi i tipi di trasporto avvengono in vescicole legate alla membrana. Queste si staccano dalla membrana del luogo da cui provengono e poi si fondono con la membrana del luogo in cui sono dirette – splendidamente raffigurato a ~2:25 nel video Life of the Cell qui sopra. Il corpo da cui si formano le vescicole è il “compartimento donatore”, e la destinazione in cui si fondono successivamente è il “compartimento accettore”.
Il processo di gemmazione richiede che le proteine G nella membrana reclutino le proteine Coat. In particolare, per il trasporto anterogrado, la proteina G Sar1 (gene: SAR1A) recluta COPII (‘cop due’); per il trasporto retrogrado, una proteina G ARF recluta COPI (pronunciato ‘cop uno’). Queste proteine G sono attivate per fare questo lavoro quando GEF le carica con GTP, sostituendo GDP.
Quindi i passi nel trasporto anterogrado, per esempio, sono i seguenti:
- Sec12-GEF (Sec sta per secretorio) carica Sar1 con GTP. Quando è legata al GDP, Sar1 galleggia semplicemente nel compartimento donatore, ma quando è legata al GTP, subisce un cambiamento conformazionale che fa sporgere la sua coda idrofoba N-terminale altrimenti sepolta, facendola aderire alla membrana, dove le proteine COPII iniziano ad accumularsi perché a loro piace molto quella coda.
- Le COPII iniziano a polimerizzare e, a causa della sua conformazione, hanno una preferenza intrinseca per la curvatura, quindi il loro accumulo inizia a far avvenire il budding. Allo stesso tempo, le proteine legate alla membrana che devono essere trasportate – identificate da una sequenza aminoacidica DXE (cioè aspartato-qualcosa-glutammato) che forma un sito di legame nella loro parte citosolica – vengono reclutate nella vescicola appena formata. Le proteine legate alla membrana agiscono come recettori, reclutando le proteine lumenali che sono legate per il Golgi per appendere fuori nello spazio concavo dove finiranno nella vescicola una volta che si forma.
- Una volta che sono arrivate abbastanza COPII, la vescicola si stacca, a questo punto Sar1 idrolizza il suo GTP, fornendo l’energia per risucchiare la sua coda idrofoba dentro di sé, tagliando le COPII. La vescicola è ora scollegata dal compartimento donatore.
- Ora, per ragioni poco spiegate (o poco comprese?), il mantello delle COPII si smonta, esponendo i recettori sotto il mantello che dirigono il puntamento della vescicola. Una volta che la vescicola arriva a destinazione, Rab-GTP incorporato nella membrana della vescicola interagisce con un effettore Rab incorporato nella membrana del compartimento accettore. Si scambia uno sguardo laterale, si accende l’interesse. Presto la vescicola si fonderà con la membrana.
- Le proteine SNARE presenti sia sulla vescicola che sulla membrana ricevente (V-SNARE e T-SNARE rispettivamente) interagiscono per avvicinare ulteriormente le membrane. In questo esempio considereremo VAMP (i geni VAMP_) come V-SNARE e Syntaxin (i geni STX__) e SNAP25 (gene SNAP25) come T-SNARE. Syntaxin e SNAP25 sono entrambe proteine di membrana; Syntaxin ha 1 elica alfa e SNAP25 ne ha 2, tutte sul lato citosolico. Le eliche alfa guidano l’interazione con VAMP. Le alfa-eliche dei lati opposti hanno un’affinità estremamente forte l’una per l’altra, portando le membrane abbastanza vicine da fondersi. Una volta che questo è successo, per staccare nuovamente le V-SNARE e le T-SNARE sono necessarie due proteine: NSF (gene: NSF; sta per NEM sensitive factor) e alfa-SNAP (gene: NAPA), una proteina solubile di attaccamento NSF. NSF è un’ATPasi, e brucia ATP per guidare il disassemblaggio energeticamente in salita del complesso.
Ora il trasporto retrogrado. Perché esiste il trasporto retrogrado? Ecco una lista non esaustiva di alcune ragioni:
- Alcune proteine di membrana iniziano la loro vita nell’ER, hanno bisogno di essere modificate nel Golgi, ma poi devono tornare nell’ER. Lo fanno con una sequenza di aminoacidi KKXX.
- C’è anche una sequenza di aminoacidi KDEL al termine C di alcune proteine lumenali che dovrebbe mantenerle nell’ER, ma non è perfetto – a volte finiscono nel Golgi, nel qual caso vengono riportate nell’ER attraverso un trasporto retrogrado che dipende dalla sequenza KDEL per il riconoscimento. Il meccanismo è abbastanza chiaro: le proteine che riconoscono e si legano a KDEL lo fanno solo a basso pH, e il pH del Golgi è più basso di quello dell’ER, quindi legano KDEL nel Golgi, poi lo rilasciano quando sono di nuovo nel pH più neutro dell’ER.
- Inoltre, pensaci, tutte le proteine che partecipano al trasporto anterogrado – le V-SNARES, Rab, ecc. – devono tornare all’ER per poter rifare tutto da capo, come l’autobus deve tornare al deposito degli autobus alla fine della giornata.
- Come vedremo tra poco, il Golgi arriva in più fasi che dipendono dall’aggiunta di enzimi più a valle.
Il processo di trasporto retrogrado non è così diverso da quello anterogrado. Utilizza ARF invece di Sar1, COPI invece di COPII, ma funziona allo stesso modo: ARF caricato con GTP lascia la sua coda idrofoba conficcarsi nella membrana, attirando l’attenzione di COPI. COPI ha due componenti, COPIalpha e COPIbeta, che interagiscono entrambi con la sequenza KKXXX per reclutare proteine legate alla membrana destinate al trasporto retrogrado. Alcune proteine hanno anche una sequenza RR (ovunque nella proteina) che può segnalarle per il trasporto retrogrado.
L’apparato del Golgi non è contiguo. È un insieme sovrapposto di sottocomparti separati chiamati sacchi o cisterne. I diversi compartimenti hanno proprietà diverse e le proteine li visitano in un ordine particolare. In ordine dall’ER alla membrana cellulare, i compartimenti del Golgi sono chiamati rete cis, mediale, trans e trans-Golgi. Ogni compartimento ha diversi enzimi che modificano le proteine, e le modifiche devono avvenire in un certo ordine, da qui la necessità di un insieme di compartimenti sovrapposti.
Ma quando le proteine maturano nel Golgi, non è come se si staccassero in vescicole da un compartimento e passassero al successivo. Piuttosto, il compartimento in cui si trovano già si muove verso l’esterno e “matura” mentre nuovi enzimi vi vengono aggiunti (da più in basso nella catena del Golgi) attraverso il trasporto retrogrado. Strano, vero? È un po’ come se, invece di passare da una scuola elementare a una scuola media a una scuola superiore, tu rimanessi in un unico edificio scolastico per tutta la tua infanzia e adolescenza, e ogni anno portassero nuovi libri di testo e nuovi insegnanti per mantenerlo adeguato al grado che tu e i tuoi compagni di classe avete ormai raggiunto. Ecco come appare il Golgi mentre si muove e si evolve:
Quindi non c’è (poco o) nessun trasporto anterogrado all’interno del Golgi, ma un sacco di trasporto retrogrado per portare ogni nuovo ciclo di enzimi. Quando le proteine hanno finalmente completato l’intero curriculum K-12 della rete del Golgi, subiscono il trasporto per passare al loro destino finale. Si staccano in una vescicola che andrà in uno dei tre posti seguenti:
- Esocitosi – fusione con la membrana cellulare. Così le proteine lumenali saranno secrete extracellularmente, e le proteine di membrana diventeranno proteine di membrana cellulare.
- Vescicole secretorie – queste rimangono come vescicole nella cellula fino a quando non sono necessarie – dove ‘necessarie’ significa che alla fine subiscono l’esocitosi. Nei neuroni, è qui che i neurotrasmettitori sono immagazzinati fino a quando un potenziale d’azione richiede la loro secrezione nella sinapsi. Nello stomaco, le cellule che producono gli enzimi gastrici conservano questi enzimi nelle vescicole secretorie fino a quando l’assunzione di cibo ne innesca il rilascio nello stomaco.
- Lisosomi – dove le proteine mal ripiegate vengono degradate.
Il trasporto dalla rete trans-Golgi a queste destinazioni è diverso dagli altri trasporti discussi sopra e spesso coinvolge la clatrina (geni CLT__). Le vescicole che si staccano hanno un rivestimento a due strati, con complessi di proteine adattatrici (AP) come strato interno e clatrina come strato esterno. Le proteine adattatrici hanno un segnale di destinazione con un motivo YXXh (h = Φ = qualsiasi amminoacido idrofobo). La clatrina forma la cosiddetta formazione ‘clatrina-triscele’ mostrata qui:

(Immagine grazie all’utente Wikimedia Commons Phoebus87)
La clatrina è anche responsabile dell’endocitosi – gemmazione di vescicole di materiale extracellulare (e proteine di membrana cellulare) per entrare nella cellula. Questo è chiamato endocitosi mediata dalla clatrina. I recettori nella membrana cellulare vengono endocitati molto frequentemente: l’intera popolazione di recettori ormonali si rinnova circa ogni ora, specialmente quando gli ormoni vengono ricevuti. Raccogliere il recettore in una vescicola è un modo per la cellula di interrompere il segnale in arrivo fino a quando non può essere elaborato.
Le note sulla membrana plasmatica discutono brevemente la fibrosi cistica: CFTR è un trasportatore ABC responsabile del pompaggio di Cl- fuori dalla cellula (lascia anche entrare Na+). I mutanti con perdita di funzione non pompano Cl-, il che rimuove la forza motrice per l’osmosi, addensando il muco e causando problemi di respirazione. Ci sono almeno 127 diversi mutanti CFTR a perdita di funzione (almeno, questo è il numero di test di Natera) che (se entrambi gli alleli sono disabilitati) causano la fibrosi cistica. La mutazione più comune è ΔF508, che è ~3% di tutti gli alleli CFTR europei e circa il 70% di quelli mutanti. La perdita di quell’unica fenilalanina cambia la conformazione di CFTR in modo che il codice di-acido di uscita (aminoacidi D565 e D567) che indirizza CFTR per le vescicole esocitotiche non è più correttamente esposto e la proteina non arriva mai alla membrana cellulare.
Sezione discussione
In sezione abbiamo letto Hu 2009, che ha dimostrato che le proteine atlastine sono coinvolte nella creazione della rete ER tubolare. La prova è venuta quasi interamente dalle interazioni proteina-proteina. Sono rimasto sorpreso che questo documento fosse un grande affare, perché ci sono stati un milione di documenti che mostrano interazioni proteina-proteina per l’huntingtina, e nessuno crede veramente a tutti loro e non ci ha necessariamente portato più vicino a sapere cosa fa l’huntingtina o cosa va storto nella malattia di Huntington. Ma apparentemente Hu è stato in grado di fare un caso abbastanza pulito per le interazioni delle atlastine con i reticoli, implicando un ruolo nella formazione dell’ER. Aiuta il fatto che Hu sia stato in grado di mostrare una “interazione genetica” oltre a un’interazione fisica (di legame). Una “interazione genetica” (ho dovuto cercarla) significa quando “A volte le mutazioni in due geni producono un fenotipo che è sorprendente alla luce degli effetti individuali di ogni mutazione. Questo fenomeno, che definisce l’interazione genetica, può rivelare relazioni funzionali tra geni e percorsi.” .
PrP
Questo è vecchio di un decennio, quindi alcune cose potrebbero essere superate, ma ho trovato la revisione di Harris 2003 (ft) sulla biologia cellulare della PrP estremamente chiara e utile. Anche Kim & Hegde 2002 è stato utile. La PrP è una proteina del percorso secretorio. I suoi primi 22 aminoacidi (MANLGCWMLVLFVATWSDLGLC) sono un peptide di segnale che causa una traslocazione cotraslazionale nell’ER. Normalmente, la PrP viene semplicemente legata a GPI al suo terminale C e viene ancorata al lato esoplasmatico della membrana. Ma gli aminoacidi 111-134 (HMAGAAAAGAVVGGLGGYMLGSAM) sono una sorta di debole sequenza di ancoraggio del segnale (Tipo II, con gli aminoacidi +++ che precedono l’ancoraggio del segnale) che a volte, ma non sempre, diventa un dominio transmembrana, invertendo il terminale C nel lume. Ancora più confuso, quella sequenza può a volte finire come un dominio transmembrana senza l’inversione, in modo che l’estremità N sia nel lume. Quindi ci sono tre topologie di membrana della PrP: la vecchia GPI-anchored, e due orientamenti transmembrana, come illustrato in Harris 2003 Fig 3:
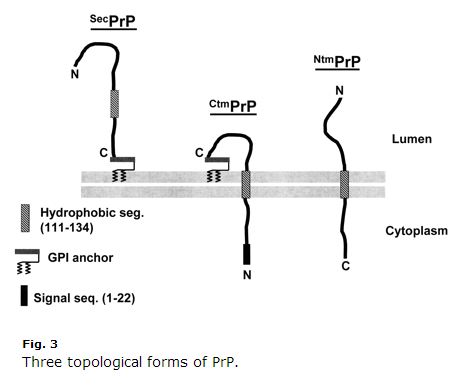
Nota come è strana la CtmPrP. È transmembrana ma anche ancorata a GPI, e il peptide di segnale N-terminale non viene mai scisso. Normalmente, le forme transmembrana sono <il 10% della PrP totale. In alcune condizioni di laboratorio la percentuale è più alta, e due delle mutazioni che causano GSS (A117V e P105L) aumentano anche la frazione di CtmPrP al 20-30% di tutta la PrP. Di queste tre forme, c’è una buona quantità di prove che la CtmPrP è tossica, e che potrebbe avere un ruolo nella formazione dei prioni, anche se la maggior parte delle mutazioni genetiche delle malattie da prioni (incluso FFI D178N) non sembrano influenzare la topologia di membrana della PrP o la frazione di CtmPrP.
Dopo che la PrP passa attraverso il Golgi, è destinata alla membrana cellulare. Ma secondo Harris, non rimane semplicemente lì – frequentemente attraverso l’endocitosi mediata dalla clatrina e attraversa la cellula ogni ~60 minuti, con alcune molecole che vengono scisse in ogni ciclo. Il rame stimola questa endocitosi di PrP. La maggior parte delle mutazioni genetiche delle malattie da prioni cambiano la localizzazione della PrP – di solito quando è presente una mutazione, meno PrP si trova sulla superficie cellulare, e più si accumula nell’ER.