Estos son los apuntes de la clase 4 del curso de Biología Celular de Harvard Extension.
La vía secretora se refiere al retículo endoplásmico, al aparato de Golgi y a las vesículas que viajan entre ellos, así como a la membrana celular y a los lisosomas. Se denomina «secretora» por ser la vía por la que la célula secreta proteínas al medio extracelular. Pero, como siempre, la etimología sólo cuenta una parte de la historia. Esta vía también procesa las proteínas que se unirán a la membrana (ya sea en la membrana celular o en las propias membranas del RE o del Golgi), así como las enzimas lisosomales, y también las proteínas que vivirán su vida en la propia vía secretora. También hace otras cosas además de procesar proteínas.
El citosol y el «lumen» (el líquido que llena la vía secretora) son ambientes químicos diferentes, y normalmente nunca se mezclan. El citosol es reductor (cuando estás en el citosol, sigues encontrándote con moléculas que quieren ofrecerte electrones), y el RE, el Golgi y el entorno extracelular son oxidativos (las moléculas siguen acercándose a ti pidiendo electrones). Consulte la sección sobre redox si sigue confundido. Esto hace que las condiciones de plegado de las proteínas sean diferentes: por ejemplo, los enlaces disulfuro suelen formarse sólo en condiciones oxidativas. Además, diferentes proteínas pueden vivir sólo en la vía secretora o sólo en el citosol. La vía secretoria proporciona una ruta para que la célula maneje cosas que no sería bueno tener en el citoplasma, y/o que son más útiles cuando se mantienen concentradas en un compartimento especializado con sus socios de interacción deseados. Los hepatocitos (en el hígado) secuestran fármacos y toxinas en el RE liso y los descomponen para excretarlos del cuerpo allí. La vía secretora no es contigua, sino que cada movimiento entre sus componentes se produce en pequeños microcosmos burbujeantes de su propio mundo químico, llamados vesículas.
Muchas proteínas que pasan por la vía secretora nunca tocan el citosol – excepto las partes de las proteínas de membrana que sobresalen en el lado citosólico. Muchas de ellas necesitan chaperonas que les ayuden a plegarse, y/o toda una serie de modificaciones postraduccionales para estar listas para su función nativa, y la vía secretora se especializa en proporcionarles todo eso.
La conferencia de hoy se centrará en cómo se traducen las proteínas en el RE y cómo viajan (en vesículas) entre el RE, el Golgi y otros destinos. Esto está muy bien representado en el vídeo La vida de la célula:
El retículo endoplásmico es el primer paso en la vía de secreción. Su membrana es continua con la membrana nuclear externa, aunque no está claro por qué eso importa, ya que no es que las proteínas comiencen su vida en el núcleo. Más bien, los ARNm vagan por el citoplasma hasta que son recogidos por un ribosoma interesado en traducirlos. En la «translocación postraduccional», la nueva proteína se traslada al RE después de ser traducida. En el fenómeno más interesante denominado «translocación cotranslacional», el ribosoma comienza la traducción como cualquier otra proteína, pero en algún punto de los primeros 16 a 30 aminoácidos encuentra un péptido señal (también conocido como secuencia señal). El motivo de la señal suele ser un aminoácido con carga positiva seguido de entre 6 y 12 aminoácidos hidrofóbicos. Este motivo es reconocido por la partícula de reconocimiento de señales (SRP, una «ribonucleoproteína» o molécula híbrida de ARN/proteína) que se une a ella e impide que el ribosoma continúe la traducción. La traducción se detiene hasta que el complejo ribosoma/SRP encuentra un receptor SRP en la membrana del RE. Cuando se encuentran, la SRP y su receptor se unen cada uno a una molécula de GTP en la membrana del RE, lo que aparentemente refuerza su interacción. Por suerte, todo esto ocurre junto a un translocon de Sec61, un complejo proteico que forma un canal que cruza la membrana del RE. El translocon es en realidad un complejo de tres proteínas diferentes (genes: SEC61A1 o SEC61A2, SEC61B, SEC61G), de las cuales la subunidad Sec61a tiene 10 a-hélices que atraviesan la membrana y forman el canal. Una vez que el ribosoma se acopla a la membrana, continúa la traducción, empujando el péptido señal y, finalmente, toda la proteína a través del canal hacia el lumen del RE. Cuando la traducción se detiene, la SRP y el receptor de la SRP hidrolizan su GTP para liberarse mutuamente y liberar la carga del ribosoma (esto tiene que requerir la energía del GTP, ya que la unión original era descendente), una peptidasa señal escinde el péptido señal de la proteína naciente, y la proteína queda libre para empezar a plegarse en el RE.
En algunas proteínas del RE intervienen un par de actores más. La oligosacárido transferasa, que añade grupos glicosilos a las asparaginas de la proteína naciente, forma parte del complejo translocon y en realidad lleva a cabo la glicosilación mientras la nueva proteína aún se está traduciendo. Así que, aunque llamemos a la glicosilación una «modificación postraduccional», en realidad se realiza durante la traducción en este caso. Además, para conseguir su estructura adecuada, algunas proteínas necesitan estar completamente traducidas antes de que se les permita empezar a plegarse; si se permitiera que la porción N-terminal empezara a plegarse tan pronto como entrara en el lumen, acabaría teniendo una estructura general incorrecta. Para evitarlo, a veces la chaperona BiP se une a la proteína para mantenerla desplegada durante un tiempo. Imagínese a BiP como otro comecocos que muerde la proteína para mantenerla lineal, como Hsc70 en el proceso de orientación mitocondrial (véase la semana pasada).
Aquí tiene un vídeo al respecto:
El primer par de minutos muestra el escenario básico descrito anteriormente. Luego pasa a un escenario más complejo que presentaré en un minuto. Para tu información, el vídeo muestra dos cosas «controvertidas» no incluidas en la descripción anterior: (1) el péptido señal que se degrada en la membrana, y (2) una «proteína tapón» que detiene el canal antes/después de la traducción. No todos los científicos están de acuerdo en estas dos cosas todavía.
Todas las proteínas que sabemos que pasan por la vía secretoria fueron señaladas allí por personas que hacían experimentos de localización para ver en qué parte de la célula se encuentra una proteína. Un hecho extraño sobre el RE es que puedes poner la célula en una licuadora y después el RE empezará a reconectarse a sí mismo, formando pequeños «microsomas» que no están unidos al núcleo sino que forman burbujas contiguas de RE. Entonces puede empezar a jugar con las proteasas -que descomponen las proteínas- y los detergentes -que solubilizan la membrana del RE-. Asumiendo que tu proteína de interés está traducida, puedes comprobar si (1) sobrevive al tratamiento con proteasas pero (2) no sobrevive al tratamiento con proteasas + detergentes, entonces es una proteína de la vía secretora. La lógica es que en el caso (1) estaba protegida dentro del RE, pero en el caso (2) disolviste el RE, por lo que fue devorada por la proteasa. Todo esto supone que tienes un anticuerpo o alguna otra forma de detectar si la proteína de interés está ahí después de estos tratamientos.
La gente también usó esas técnicas para averiguar que sólo 70 aminoácidos de una nueva proteína pueden ser traducidos antes de que sea demasiado tarde para que esa proteína acabe en el RE. Recuerde, el péptido señal está en los primeros 16-30 aminoácidos, y la translocación al RE depende de que el SRP esté presente. Los ribosomas traducen a un ritmo predecible, por lo que la gente hizo que los ribosomas empezaran a traducir algo de ARNm y luego esperaron cantidades de tiempo determinadas antes de añadir SRP, para ver cuánta traducción podía ocurrir antes de que el SRP ya no pudiera hacer su trabajo.
El receptor SRP y las proteínas Sec61 son proteínas de la membrana del RE – y hay muchas otras proteínas de la membrana del RE, de la membrana del Golgi y de la membrana del lisosoma también. De hecho, incluso las proteínas de membrana (ver clase 02) de la membrana celular se procesan en la vía secretora. Muchas de ellas tienen varios o decenas de dominios transmembrana (20-25 aminoácidos hidrofóbicos cada uno) que tienen que ser insertados en el orden y la orientación correctos (por ejemplo, realmente quieres que tus canales de iones y transportadores apunten en la dirección correcta, hacia dentro o hacia fuera de la célula). En consecuencia, hay un montón de mecanismos biológicos extravagantes para conseguir que estas proteínas se inserten correctamente en la membrana. Esto es lo que muestra la última mitad del vídeo anterior.
Así que aquí hay una tautología: algunas proteínas tienen una secuencia topogénica que determina su orientación en la membrana. Esta secuencia se compone de dos tipos de secuencias de señal:
- una secuencia de parada-transferencia (abreviada STA por alguna razón) es una secuencia de 22-25 aminoácidos hidrofóbicos en algún lugar en el medio de la proteína que forma una hélice alfa. Cuando se encuentra es empujada hacia la membrana, y entonces la traducción del resto de la proteína continúa en el citosol. Así que esto como que «deshace» la translocación al RE que fue iniciada por el péptido señal al principio (terminación N) de la proteína.
- una secuencia de anclaje de señal (abreviada SA) es también una hélice alfa hidrofóbica de 22-25aa, pero con una serie de ~3 aminoácidos cargados positivamente a su izquierda o derecha. Al igual que el péptido señal, es reconocido por la SRP, que lleva el ribosoma al RE. Pero a diferencia del péptido señal, esta secuencia alfa helicoidal se insertará en la membrana del RE. La orientación de la inserción está determinada por los 3 aminoácidos con carga positiva. Las cargas positivas tienen que terminar siempre en el lado citosólico, por lo que si vienen después (es decir, C-terminal de) la secuencia hidrofóbica, la proteína termina con su extremo terminal C apuntando hacia el citosol, pero si vienen antes (es decir, N-terminal de) la secuencia hidrofóbica, la proteína termina con su extremo N apuntando hacia el citosol.
Con esas dos señales como bloques de construcción, se puede imaginar una proteína con una serie de secuencias de transferencia de parada y de anclaje de señales para crear toda una serie de dominios transmembrana de ida y vuelta cosidos en la membrana como por una máquina de coser. La gente ha clasificado las proteínas de membrana en cinco categorías:
- El tipo I tiene sólo un péptido señal y luego una transferencia de parada en el medio. Por lo tanto, termina con su extremo N (hidrofílico) en el lumen, su medio (hidrofóbico) en la membrana y su extremo C (hidrofílico) en el citosol.
- El tipo II no comienza con un péptido señal. Comienza como cualquier otra proteína, pero en el medio tiene una secuencia de anclaje de señal con los aminoácidos +++ primero y la serie hidrofóbica después. Esto hace que la proteína se transloque a mitad de la traducción, con la parte N-terminal ya traducida sobresaliendo en el citosol (ya que los +++ tienen que permanecer en el citosol) y la parte C-terminal que ahora empieza a ser traducida se traduce directamente en el RE. Así que termina transmembrana con su C-terminal en el ER y N-terminal en el citosol – opuesto al Tipo I.
- El Tipo III es como el Tipo II – sin péptido de señal, sólo un ancla de señal en el medio, pero en este caso los +++ vienen después de la secuencia hidrofóbica, que invierte la orientación. Así que termina con su extremo N en el RE y su extremo C en el citosol. Lo contrario del Tipo II y, al final, lo mismo que el Tipo I, aunque llegó allí de una manera diferente – no tiene un péptido señal que se escinde en el RE.
- Las proteínas del Tipo IV o «multipasos» tienen una serie alterna de secuencias señal y secuencias de transferencia de parada. Estas son claramente más de un «tipo», pero no son tan diversas como su imaginación combinatoria podría permitir. La orientación de la primera secuencia señal determina si la terminación N acabará en el citosol o en el RE, y el número total de secuencias de transferencia de parada + anclaje de señal determina dónde acabará la terminación C: un número par = el mismo lado que la terminación N, un número impar = el lado opuesto a la terminación N. Las secuencias STA y SA tienen que alternarse estrictamente, con la excepción de que se puede empezar con dos secuencias de anclaje de señal si la primera está orientada con la terminación N hacia el citosol. Sólo para burlarse de este esquema de categorización, la gente ha definido algunos subtipos incompletos del Tipo IV, donde el Tipo IVa es N-terminal en el citosol (por lo tanto comienza como una proteína del Tipo II) y el Tipo IVb es N-terminal en el lumen (comienza como una proteína del Tipo III pero luego tiene otra secuencia SA que la devuelve al RE). GLUT1 de la clase 02 es una Tipo IVa.
- Las proteínas ancladas a la GPI, que son el quinto tipo pero no se llaman Tipo V, comienzan con un péptido señal y terminan con un C-terminal hidrofóbico que permanece incrustado en la membrana. Ese extremo hidrofóbico se escinde y se sustituye por GPI, que también permanece incrustado en la membrana. La PrP es una de ellas – más adelante hablaremos de ello.
Ahora ya hemos hablado de cómo las proteínas pueden acabar en el lumen del RE o atravesando la membrana del RE. La mayoría de las proteínas abandonan el RE en cuestión de minutos, transportadas en vesículas con destino al Golgi y posteriormente a la excreción, a los lisosomas o a la membrana celular. Esa dirección de viaje hacia delante se denomina anterógrada; ir hacia atrás desde el Golgi al RE es un transporte retrógrado.
Ambos tipos de transporte tienen lugar en vesículas unidas a la membrana. Éstas se desprenden de la membrana del lugar del que proceden y, posteriormente, se fusionan con la membrana del lugar al que se dirigen, tal y como se muestra en el minuto 2:25 del vídeo sobre la vida de la célula. El cuerpo del que se forman las vesículas es el «compartimento donante», y el destino al que posteriormente se fusionan es el «compartimento receptor».
El proceso de brotación requiere que las proteínas G de la membrana recluten a las proteínas Coat. En concreto, para el transporte anterógrado, la proteína G Sar1 (gen: SAR1A) recluta a COPII (‘cop dos’); para el transporte retrógrado, una proteína G ARF recluta a COPI (pronunciado ‘cop uno’). Estas proteínas G se activan para hacer este trabajo cuando el GEF las carga con GTP, intercambiando el GDP.
Así que los pasos en el transporte anterógrado, por ejemplo, son los siguientes:
- Sec12-GEF (Sec significa secretor) carga Sar1 con GTP. Cuando se une a GDP, Sar1 simplemente flota alrededor del compartimento donante, pero cuando se une a GTP, sufre un cambio conformacional que hace que su cola hidrofóbica N-terminal, que de otro modo estaría quemada, sobresalga, haciendo que se pegue a la membrana, donde las proteínas COPII comienzan entonces a acumularse porque les gusta mucho esa cola.
- Las COPIIs comienzan a polimerizarse y, debido a su conformación, tienen una preferencia intrínseca por la curvatura, por lo que su acumulación comienza a hacer que se produzca la gemación. Al mismo tiempo, las proteínas unidas a la membrana que necesitan ser transportadas -identificadas por una secuencia de aminoácidos DXE (es decir, aspartato-glutamato) que forma un sitio de unión en su parte citosólica- son reclutadas a la vesícula recién formada. Las proteínas unidas a la membrana actúan como receptores, reclutando a las proteínas del lumen que están unidas al Golgi para que se queden en el espacio cóncavo donde acabarán en la vesícula una vez que ésta se forme.
- Una vez que han llegado suficientes COPIIs, la vesícula se desprende, momento en el que Sar1 hidroliza su GTP, proporcionando la energía para que succione su cola hidrofóbica de vuelta a sí misma, cortando los COPIIs. La vesícula está ahora desconectada del compartimento donante.
- Ahora, por razones mal explicadas (¿o mal entendidas?), la capa de los COPIIs se acaba de desmontar, exponiendo receptores bajo la capa que dirigen la orientación de la vesícula. Una vez que la vesícula llega a su destino, el Rab-GTP incrustado en la membrana de la vesícula interactúa con un efector Rab incrustado en la membrana del compartimento aceptor. Se intercambia una mirada de reojo y se despierta el interés. Pronto la vesícula se fusionará con la membrana.
- Las proteínas SNARE presentes tanto en la vesícula como en la membrana receptora (V-SNARE y T-SNARE respectivamente) interactúan para acercar aún más las membranas. En este ejemplo consideraremos a VAMP (los genes VAMP_) como las V-SNARE y a Syntaxin (los genes STX__) y SNAP25 (el gen SNAP25) como las T-SNARE. Syntaxin y SNAP25 son proteínas de membrana; Syntaxin tiene 1 hélice alfa y SNAP25 tiene 2, todas en el lado citosólico. Las hélices alfa impulsan la interacción con VAMP. Las hélices alfa de los lados opuestos tienen una afinidad extremadamente fuerte entre sí, acercando las membranas lo suficiente como para fusionarse. Una vez que esto ha ocurrido, separar las V-SNAREs y las T-SNAREs requiere dos proteínas: La NSF (gen: NSF; significa factor sensible a la NEM) y la alfa-SNAP (gen: NAPA), una proteína soluble de unión a la NSF. NSF es una ATPasa, y quema ATP para impulsar el desensamblaje energéticamente ascendente del complejo.
Ahora el transporte retrógrado. ¿Por qué hay el transporte retrógrado en absoluto? Aquí hay una lista no exhaustiva de algunas razones:
- Algunas proteínas de membrana comienzan su vida en el RE, necesitan modificarse en el Golgi, pero luego necesitan volver al RE. Lo hacen con una secuencia de aminoácidos KKXX.
- También hay una secuencia de aminoácidos KDEL en la terminación C de algunas proteínas lumenales que se supone que las mantiene en el RE, pero no es perfecto – a veces acaban en el Golgi, en cuyo caso se dirigen de nuevo al RE mediante un transporte retrógrado que depende de esa secuencia KDEL para su reconocimiento. El mecanismo es bastante claro – las proteínas que reconocen y se unen a KDEL sólo lo hacen a un pH bajo, y el pH del Golgi es más bajo que el del RE, así que se unen a KDEL en el Golgi, y luego lo liberan cuando están de vuelta en el pH más neutro del RE.
- Además, piénsalo, todas las proteínas que participan en el transporte anterógrado – las V-SNARES, Rab, etc. – tienen que volver al RE para poder hacerlo de nuevo, como el autobús tiene que volver a la estación de autobuses al final del día.
- Como veremos en breve, el Golgi viene en múltiples etapas que dependen de la adición de enzimas de más abajo.
El proceso de transporte retrógrado no es tan diferente del anterógrado. Utiliza ARF en lugar de Sar1, COPI en lugar de COPII, pero funciona igual: ARF cargado con GTP deja que su cola hidrofóbica se adhiera a la membrana, atrayendo la atención de COPI. La COPI tiene dos componentes, la COPIalfa y la COPIbeta, que interactúan con esa secuencia KKXXX para reclutar proteínas unidas a la membrana destinadas al transporte retrógrado. Algunas proteínas también tienen una secuencia RR (en cualquier parte de la proteína) que puede marcarlas para el transporte retrógrado.
El aparato de Golgi no es contiguo. Es un conjunto apilado de subcompartimentos separados llamados sacos o cisternas. Los distintos compartimentos tienen propiedades diferentes y las proteínas los visitan en un orden determinado. En orden desde el RE a la membrana celular, los compartimentos de Golgi se denominan red cis, medial, trans y trans-Golgi. Cada compartimento tiene diferentes enzimas que modifican las proteínas, y las modificaciones tienen que producirse en un orden determinado, de ahí la necesidad de un conjunto apilado de compartimentos.
Pero a medida que las proteínas maduran en el Golgi, no es que broten en vesículas de un compartimento y se trasladen al siguiente. Más bien, el compartimento en el que ya se encuentran se desplaza hacia fuera y «madura» a medida que se le añaden nuevas enzimas (desde más abajo en la cadena de Golgi) a través del transporte retrógrado. Extraño, ¿verdad? Es como si, en lugar de pasar de la escuela primaria a la secundaria, te quedaras en un mismo edificio escolar durante toda tu infancia y adolescencia, y simplemente trajeran nuevos libros de texto y profesores cada año para mantenerlo apropiado para el grado que tú y tus compañeros habéis alcanzado. Este es el aspecto del Golgi cuando se mueve y evoluciona:
Así que hay (poco o) ningún transporte anterógrado dentro del Golgi, pero mucho transporte retrógrado para traer cada nueva ronda de enzimas. Cuando las proteínas han completado el plan de estudios K-12 de la red de Golgi, son transportadas a su destino final. Salen en una vesícula que irá a uno de estos tres lugares:
- Exocitosis – fusión con la membrana celular. Así, las proteínas del lumen serán secretadas extracelularmente, y las proteínas de la membrana se convertirán en proteínas de la membrana celular.
- Vesículas secretoras – se quedan como vesículas en la célula hasta que se necesiten – donde «se necesitan» significa que eventualmente sufren exocitosis. En las neuronas, es donde se almacenan los neurotransmisores hasta que un potencial de acción exige su secreción en la sinapsis. En el estómago, las células que producen enzimas gástricas las guardan en vesículas secretoras hasta que la ingesta de alimentos desencadena su liberación en el estómago.
- Lisosomas – donde van las proteínas mal plegadas para ser degradadas.
El transporte desde la red trans-Golgi a estos destinos es diferente del otro transporte discutido anteriormente y a menudo implica a la clatrina (genes CLT__). Las vesículas que brotan tienen una cubierta de dos capas, con complejos de proteínas adaptadoras (AP) como capa interna y clatrina como capa externa. Las proteínas adaptadoras tienen una señal de destino con un motivo YXXh (h = Φ = cualquier aminoácido hidrofóbico). La clatrina forma la llamada «formación de clatrina-triskelion» que se muestra aquí:

(Imagen gracias al usuario de Wikimedia Commons Phoebus87)
La clatrina también es responsable de la endocitosis, es decir, del brote de vesículas de material extracelular (y proteínas de la membrana celular) para entrar en la célula. Esto se llama endocitosis mediada por clatrina. Los receptores de la membrana celular se endocitan con mucha frecuencia: toda la población de receptores hormonales cambia aproximadamente cada hora, especialmente cuando se reciben hormonas. Llevar el receptor a una vesícula es una forma de que la célula corte la señal entrante hasta que pueda ser procesada.
Las notas sobre la membrana plasmática tratan brevemente la fibrosis quística: El CFTR es un transportador ABC responsable de bombear Cl- fuera de la célula (también deja entrar Na+). Los mutantes con pérdida de función no bombean Cl-, lo que elimina la fuerza motriz de la ósmosis, espesando el moco y causando problemas respiratorios. Hay al menos 127 mutantes de pérdida de función de CFTR diferentes (al menos, ese es el número de pruebas de Natera) que (si ambos alelos están desactivados) causan fibrosis quística. La mutación más común es la ΔF508, que representa el ~3% de todos los alelos CFTR europeos y alrededor del 70% de los mutantes. La pérdida de esa única fenilalanina cambia la conformación de CFTR de modo que el código de salida diácido (aminoácidos D565 y D567) que dirige a CFTR hacia las vesículas exocitóticas ya no está correctamente expuesto y la proteína nunca llega a la membrana celular.
Sección de discusión
En la sección leemos a Hu 2009, que demostró que las proteínas atlastin están implicadas en la creación de la red tubular de ER. La evidencia vino casi en su totalidad de las interacciones proteína-proteína. Me sorprendió que este artículo fuera algo importante, porque ha habido un millón de artículos que muestran interacciones proteína-proteína para la huntingtina, y nadie se los cree realmente todos y no nos ha acercado necesariamente a saber qué hace la huntingtina o qué va mal en la enfermedad de Huntington. Pero, aparentemente, Hu ha sido capaz de demostrar que las interacciones de las atlastinas con los reticulones implican un papel en la formación del RE. Ayuda el hecho de que Hu fuera capaz de mostrar una «interacción genética» además de una interacción física (de unión). Una ‘interacción genética’ (he tenido que buscarlo) significa cuando «A veces, las mutaciones en dos genes producen un fenotipo que es sorprendente a la luz de los efectos individuales de cada mutación. Este fenómeno, que define la interacción genética, puede revelar relaciones funcionales entre genes y vías.» .
PrP
Esto es de hace una década, así que algunas cosas pueden estar anticuadas, pero encontré la revisión de Harris 2003 (ft) sobre la biología celular de la PrP extremadamente clara y útil. Kim & Hegde 2002 también fue útil. La PrP es una proteína de la vía secretora. Sus primeros 22 aminoácidos (MANLGCWMLVLFVATWSDLGLC) son un péptido señal que provoca la translocación cotranslacional al RE. Normalmente, la PrP sólo se une a la GPI en su extremo C y se ancla al lado exoplásmico de la membrana. Pero los aminoácidos 111-134 (HMAGAAAAGAVVGGLGGYMLGSAM) son una especie de secuencia de anclaje de señal débil (Tipo II, con los aminoácidos +++ que vienen antes del anclaje de señal) que a veces, pero no siempre, se convierte en un dominio transmembrana, invirtiendo el extremo C en el lumen. Y lo que es más confuso, a veces esa secuencia puede terminar como un dominio transmembrana sin la inversión, de modo que el extremo N está en el lumen. Así que hay tres topologías de membrana de la PrP: la antigua anclada a la GPI, y dos orientaciones transmembrana, como se muestra en la Fig. 3 de Harris 2003:
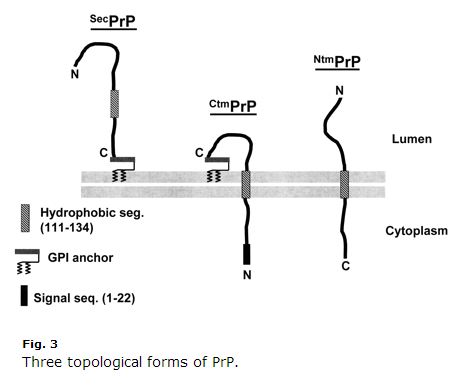
Nótese lo extraña que es la CtmPrP. Es transmembrana pero también anclada a GPI, y el péptido señal N-terminal nunca se escinde. Normalmente, las formas transmembrana son < el 10% de la PrP total. En algunas condiciones de laboratorio el porcentaje es mayor, y dos de las mutaciones que causan GSS (A117V y P105L) también aumentan la fracción de CtmPrP al 20-30% de toda la PrP. De estas tres formas, hay una buena cantidad de pruebas de que la CtmPrP es tóxica, y de que podría desempeñar un papel en la formación de priones, aunque la mayoría de las mutaciones genéticas de la enfermedad priónica (incluido el FFI D178N) no parecen afectar a la topología de la membrana de la PrP o a la fracción de CtmPrP.
Después de que la PrP pase por el Golgi, se dirige a la membrana celular. Pero según Harris, no se queda ahí, sino que con frecuencia pasa por la endocitosis mediada por clatrina y hace un ciclo a través de la célula cada ~60 minutos, en el que algunas moléculas se escinden en cada ciclo. El cobre estimula esta endocitosis de la PrP. La mayoría de las mutaciones genéticas de las enfermedades priónicas cambian la localización de la PrP: normalmente, cuando hay una mutación, se encuentra menos PrP en la superficie celular y se acumula más en el RE.