Estas são notas da palestra 4 do curso de Biologia Celular de Harvard Extension.
O caminho secreto refere-se ao retículo endoplasmático, aparelho de Golgi e as vesículas que viajam entre eles, bem como a membrana celular e lisossomos. É chamado ‘secretory’ por ser o caminho pelo qual a célula segrega as proteínas para o ambiente extracelular. Mas como sempre, a etimologia só conta uma fração da história. Este caminho também processa proteínas que serão ligadas à membrana (seja na membrana celular ou nas próprias membranas de ER ou Golgi), assim como enzimas lisossômicas, e também quaisquer proteínas que viverão suas vidas no próprio caminho secreto. Ele também faz algumas coisas além de processar proteínas.
O citosol e o ‘lúmen’ (o líquido que preenche o caminho secreto) são ambientes químicos diferentes, e eles normalmente nunca se misturam. O citosol é redutor (quando você está no citosol, você continua encontrando moléculas que querem lhe oferecer elétrons), e as ER, Golgi e ambiente extracelular são oxidantes (moléculas continuam vindo até você pedindo por elétrons). Veja redox se ainda estiver confuso. Isto faz com que haja diferentes condições de dobramento de proteínas: por exemplo, as ligações de dissulfeto geralmente só se formam em condições oxidativas. Além disso, proteínas diferentes podem viver apenas na via secreta ou apenas no citosol. A via secreta fornece uma rota para a célula lidar com coisas que podem não ser boas de se ter no citoplasma, e/ou são mais úteis quando mantidas concentradas em um compartimento especializado com seus parceiros interativos desejados. Os hepatócitos (no fígado) sequestram drogas e toxinas nas Urgências lisas e decompõem-nas para excreção do corpo. A via secreta não é contígua, mas cada movimento entre os seus componentes está em pequenos microcosmos borbulhantes do seu próprio mundo químico, chamados vesículas.
Muitas proteínas que passam pela via secreta nunca tocam no citosol – excepto as partes das proteínas da membrana que se destacam no lado citosólico. Muitas delas precisam de chaperones para ajudar na dobra, e/ou uma série de modificações pós-tradução para estarem prontas para sua função nativa, e o caminho secreto é especializado em fornecer-lhes tudo isso.
A palestra de hoje vai focar em como as proteínas são traduzidas para as ER e como elas viajam (em vesículas) entre as ER, Golgi e outros destinos. Isto é belamente retratado no vídeo Vida da Célula:
O retículo endoplasmático é o primeiro passo no caminho do segredo. Sua membrana é contínua com a membrana nuclear externa, embora não esteja claro por que isso importa, já que não é como as proteínas que começam sua vida no núcleo. Pelo contrário, os mRNAs andam à deriva no citoplasma até serem capturados por um ribossomo interessado em traduzi-los. Na ‘translocação pós-tradicional’ a nova proteína é movida para as urgências após ser traduzida. No fenómeno mais interessante chamado ‘translocação cotradlacional’ o ribossoma começa a traduzir como qualquer outra proteína, mas algures nos primeiros 16 a 30 aminoácidos atinge um peptídeo de sinal (também conhecido como sequência de sinal). O motivo desse sinal é frequentemente 1 aminoácido com carga positiva seguido por 6-12 aminoácidos hidrófobos. Este motivo é reconhecido pela partícula de reconhecimento do sinal (SRP, uma ‘ribonucleoproteína’ ou molécula híbrida de RNA/proteína) que se liga a ele e impede o ribossomo de continuar a tradução. A tradução é interrompida até o complexo ribossomo/SRP encontrar um receptor SRP na membrana do ER. Quando se encontram, o SRP e o seu receptor ligam-se cada um a uma molécula GTP na membrana do ER, o que aparentemente reforça a sua interacção. Fortuitamente, tudo isto acontece adjacente a um Sec61 translocon – um complexo proteico que forma um canal que atravessa a membrana do ER. O translocon é na verdade um complexo de três proteínas diferentes (genes: SEC61A1 ou SEC61A2, SEC61B, SEC61G), das quais a subunidade Sec61a tem 10 membranas que formam o canal. Uma vez atracado o ribossomo na membrana, continua a translação, empurrando o peptídeo de sinal e eventualmente toda a proteína através do canal para o lúmen das ER. Quando a translação pára, o SRP e o receptor SRP hidrolzam o seu GTP para se libertarem mutuamente e a carga do ribossoma (isto tem de requerer a energia do GTP, uma vez que a ligação original era descendente), uma peptidase de sinal separa o peptídeo de sinal da proteína nascente, e a proteína é livre para começar a dobrar no ER.
Alguns outros jogadores estão envolvidos para algumas proteínas do ER. A oligossacarídeo transferase, que adiciona grupos glicosil às asparaginas na proteína nascente, faz parte do complexo translocon e, na verdade, realiza a glicosilação enquanto a nova proteína ainda está sendo traduzida. Portanto, embora chamemos a glicosilação de “modificação pós-tradução”, ela é realmente feita durante a tradução, neste caso. Além disso, para atingir sua estrutura adequada, algumas proteínas precisam ser totalmente traduzidas antes de poderem começar a dobrar – se a porção N-terminal fosse autorizada a começar a dobrar assim que entrasse no lúmen, acabaria com a estrutura geral errada. Para evitar isso, às vezes o BiP o acompanhante liga a proteína para mantê-la desdobrada por algum tempo. Imagine o BiP como outro Pac-Man que morde a proteína para mantê-la linear, como o Hsc70 no processo de mira mitocondrial (ver semana passada).
Aqui está um vídeo dele:
Os primeiros minutos mostram o cenário básico descrito acima. Depois ele passa para um cenário mais complexo que vou apresentar em um minuto. FYI, o vídeo mostra duas coisas ‘controversas’ não incluídas na descrição acima: (1) o peptídeo de sinal sendo degradado na membrana, e (2) uma ‘plug protein’ que pára o canal antes/depois da tradução. Nem todos os cientistas concordam ainda sobre estas duas coisas.
Todas as proteínas que sabemos que passam pelo caminho secreto foram identificadas lá por pessoas fazendo experimentos de localização para ver onde está uma proteína na célula. Um fato estranho sobre o ER é que você pode colocar a célula em um liquidificador e depois o ER vai começar a se reconectar a si mesmo, formando pequenos ‘microsomos’ que não estão ligados ao núcleo, mas formam bolhas contíguas de ER. Pode então começar a jogar jogos com proteases – que decompõem proteínas – e detergentes – que solubilizam a membrana do ER. Assumindo que a sua proteína de interesse é traduzida, pode verificar se ela (1) sobrevive ao tratamento com proteases mas (2) não sobrevive ao tratamento com proteases + detergentes, então é uma proteína do caminho secreto. A lógica é que no caso (1) ela foi protegida dentro da ER, mas no caso (2) você dissolveu a ER, então ela foi comida pela protease. Tudo isto pressupõe que tem um anticorpo ou outra forma de detectar se a proteína de interesse está lá depois destes tratamentos.
As pessoas também utilizaram estas técnicas para descobrir que apenas 70 aminoácidos de uma nova proteína podem ser traduzidos antes que seja tarde demais para que essa proteína acabe no ER. Lembre-se, o peptídeo de sinal está nos primeiros 16-30 aminoácidos, e a translocação para o ER depende da presença do SRP. Os ribossomos traduzem a uma taxa previsível, então as pessoas começaram a traduzir alguns mRNA e depois esperaram um certo tempo antes de adicionar o SRP, para ver o quanto de tradução poderia ocorrer antes que o SRP não pudesse mais fazer seu trabalho.
O receptor SRP e as proteínas Sec61 são proteínas da membrana do ER – e há muitas outras proteínas da membrana do ER, membrana de Golgi e proteínas da membrana do lisossomo também. Na verdade, mesmo as proteínas da membrana (ver classe 02) da membrana celular são processadas na via secreta. Muitas delas têm vários ou dezenas de domínios transmembrana (20-25 aminoácidos hidrofóbicos cada) que têm de ser inseridos na ordem e orientação correctas (por exemplo, você quer realmente que os seus canais iónicos e transportadores estejam apontados na direcção correcta, dentro vs. fora da célula). Por conseguinte, há um monte de mecanismos biológicos sofisticados para que estas proteínas sejam inseridas correctamente na membrana. Isto é o que a última metade do vídeo acima retrata.
Então aqui está uma tautologia: algumas proteínas têm uma sequência topogénica que determina a sua orientação na membrana. Esta sequência é feita de dois tipos de sequências de sinal:
- uma sequência de stop-transferência (STA abreviada por alguma razão) é uma sequência de 22-25 aminoácidos hidrofóbicos algures no meio da proteína que forma uma hélice alfa. Quando encontrada é empurrada para dentro da membrana, e então a tradução do resto da proteína continua no citosol. Então este tipo de ‘desfaz’ a translocação para a ER que foi iniciada pelo peptídeo de sinal no início (terminal N) da proteína.
- a sequência de ancoragem do sinal (abreviada SA) também é uma hélice alfa hidrofóbica 22-25aa, mas com uma série de ~3 aminoácidos com carga positiva à sua esquerda ou direita. Tal como o peptídeo de sinal, este é reconhecido pela SRP, que traz o ribossoma para as Urgências. Mas, ao contrário do peptídeo de sinal, esta sequência alfa helicoidal será inserida na membrana do ER. A orientação da inserção é determinada pelos 3 aminoácidos com carga positiva. As cargas positivas têm de acabar sempre no lado citosólico, portanto se vierem depois (i.e. C-terminal da) sequência hidrofóbica, a proteína acaba com a sua extremidade terminal C apontada para o citosol, mas se vierem antes (i.e. N-terminal da) sequência hidrofóbica, a proteína acaba com a sua extremidade N apontada para o citosol.
Com esses dois sinais como blocos de construção, você pode imaginar uma proteína com uma série de seqüências de transferência de stop e âncora de sinal para criar uma série inteira de domínios transmembrana para frente e para trás costurados na membrana como se fosse por uma máquina de costura. As pessoas classificaram as proteínas da membrana em cinco categorias:
- Type I tem apenas um peptídeo de sinal e depois uma paragem de transferência no meio. Portanto acaba com o seu terminal (hidrofílico) N no lúmen, o seu meio (hidrofóbico) na membrana e o seu terminal (hidrofílico) C no citosol.
- Type II não começa com um peptídeo de sinal. Começa como qualquer outra proteína, mas no meio tem uma sequência de âncora de sinal com os +++ aminoácidos vindo primeiro e a série hidrofóbica depois. Isto faz com que a proteína seja translocada a meio da tradução, com a parte terminal N já traduzida para o citosol (uma vez que o +++ tem de se manter citosólico) e a parte terminal C, agora em início de tradução, a ser traduzida directamente para as Urgências. Então ela acaba transmembrana com seu terminal C no ER e terminal N no citosol – oposto do Tipo I.
- Tipo III é como o Tipo II – sem peptídeo de sinal, apenas uma âncora de sinal no meio, mas neste caso o ++++ vem após a seqüência hidrofóbica, o que reverte a orientação. Então isto acaba com seu terminal N no ER e seu terminal C no citosol. Ao contrário do Tipo II e, no final, o mesmo que o Tipo I, embora tenha chegado lá de uma forma diferente – não tem um peptídeo de sinal que é clivado no ER.
- Proteínas do Tipo IV ou ‘multipass’ têm uma série alternada de seqüências de sinal e de transferência de stop. Estas são claramente mais do que um ‘tipo’, mas não são tão diversas como a sua imaginação combinatória poderia permitir. A orientação da primeira sequência de sinal determina se o terminal N terminará no citosol ou ER, e o número total de sequências de paragem de transferência + âncora de sinal determina onde o terminal C terminará: um número par = mesmo lado que o terminal N, número ímpar = lado oposto ao terminal N. As sequências STA e SA têm que se alternar estritamente, com a exceção de que você pode começar com duas sequências de âncora de sinal se a primeira estiver orientada com o terminal N para o citosol. Apenas para fazer um escárnio deste esquema de categorização, as pessoas definiram alguns subtipos incompletamente definidos de Tipo IV, onde Tipo IVa é N-terminal no citosol (assim começa como uma proteína Tipo II) e Tipo IVb é N-terminal no lúmen (começa como uma proteína Tipo III mas depois tem outra sequência SA que a coloca de volta no ER). GLUT1 da Classe 02 é uma proteína Tipo IVa.
- proteínas GPI-anchored, que são o quinto tipo mas não são chamadas de Tipo V, começam com um peptídeo de sinal e terminam com um termo C hidrofóbico que fica embutido na membrana. Essa extremidade hidrofóbica é clivada e substituída por GPI, que também permanece incrustada na membrana. PrP é um destes – mais sobre isso mais tarde.
Até agora já discutimos como as proteínas podem acabar no lúmen do ER ou atravessar a membrana do ER. A maioria das proteínas deixa o ER em minutos, transportadas em vesículas destinadas ao Golgi e depois para excreção, lisossomas ou a membrana celular. Essa direção de viagem para frente é chamada de anterógrada; ir para trás do Golgi para o ER é transporte retrógrado.
Bambos os tipos de transporte ocorrem em vesículas ligadas à membrana. Estes brotam da membrana de onde quer que venham, e mais tarde se fundem com a membrana de onde quer que se dirijam – lindamente retratado a ~2:25 no vídeo da Vida da Célula acima. O corpo do qual as vesículas se formam é o ‘compartimento doador’, e o destino para o qual se fundem mais tarde é o ‘compartimento aceitador’.
O processo de formação do rebento requer que as proteínas G na membrana recrutem as proteínas do revestimento. Especificamente, para o transporte anterógrado, a proteína G Sar1 (gene: SAR1A) recruta COPII (‘cop two’); para o transporte retrógrado, uma proteína ARF G recruta COPI (pronuncia-se ‘cop one’). Estas proteínas G são ativadas para fazer este trabalho quando GEF as carrega com GTP, trocando o GDP.
Então os passos no transporte anterógrado, por exemplo, são os seguintes:
- Sec12-GEF (Sec significa secretory) carrega Sar1 com GTP. Quando ligado ao GDP, o Sar1 apenas flutua ao redor do compartimento doador, mas quando ligado ao GTP, ele sofre uma mudança conformacional que faz com que sua outra cauda N-terminal hidrofóbica enterrada se projete, fazendo com que ela se cole na membrana, onde as proteínas COPII então começam a acumular porque elas realmente gostam daquela cauda.
- Os COPIIs começam a polimerizar e, devido a sua conformação, têm uma preferência intrínseca pela curvatura, de modo que seu acúmulo começa a fazer com que a brotação aconteça. Ao mesmo tempo, proteínas ligadas à membrana que precisam ser transportadas – identificadas por uma sequência de aminoácidos DXE (ou seja, aspartato – qualquer coisa-glutamato) que forma um local de ligação na sua parte citosólica – são recrutadas para a vesícula recém-formada. As proteínas ligadas à membrana atuam como receptores, recrutando proteínas lumenais que são ligadas para que o Golgi fique no espaço côncavo, onde acabarão na vesícula uma vez formadas.
- Após a chegada dos COPII suficientes, os botões da vesícula, a Sar1 hidrolisa o seu GTP, fornecendo a energia para sugar a sua cauda hidrofóbica de volta para dentro de si, cortando os COPII soltos. A vesícula está agora desligada do compartimento doador.
- Agora, por razões mal explicadas (ou mal compreendidas?), a pelagem dos COPIIs apenas se desmonta, expondo receptores sob a pelagem que direcionam o alvo da vesícula. Uma vez que a vesícula chega ao seu destino, Rab-GTP embutido na membrana da vesícula interage com um efeito Rab embutido na membrana do compartimento de aceitação. Um olhar lateral é trocado, o interesse é despertado. Logo a vesícula se fundirá com a membrana.
- proteínas SNARE presentes tanto na vesícula quanto na membrana alvo (V-SNARE e T-SNARE respectivamente) interagem para aproximar ainda mais as membranas. Neste exemplo vamos considerar VAMP (os genes VAMP_) como os genes V-SNARE e Syntaxin (os genes STX__) e SNAP25 (o gene SNAP25) como os T-SNAREs. A Syntaxin e SNAP25 são ambas proteínas de membrana; a Syntaxin tem 1 hélice alfa e a SNAP25 tem 2, todas no lado citosólico. As hélices alfa impulsionam a interação com o VAMP. As hélices alfa dos lados opostos têm uma afinidade extremamente forte umas com as outras, aproximando as membranas o suficiente para se fundirem. Uma vez que isso tenha acontecido, para separar novamente os V-SNAREs e T-SNAREs, são necessárias duas proteínas: NSF (gene: NSF; significa factor sensível NEM) e alfa-SNAP (gene: NAPA), uma proteína solúvel de fixação NSF. NSF é uma ATPase, e queima ATP para impulsionar a desmontagem energética do complexo.
Agora para transporte retrógrado. Porque é que existe transporte retrógrado? Aqui está uma lista não exaustiva de algumas razões:
- algumas proteínas de membrana começam a sua vida no ER, precisam de ser modificadas no Golgi, mas depois precisam de voltar para o ER. Eles fazem isso com uma sequência de aminoácidos KKXX.
- Existe também uma sequência de aminoácidos KDEL no terminal C de algumas proteínas lumenais que é suposto mantê-las no ER, mas não é perfeito – às vezes elas acabam no Golgi, e nesse caso são direccionadas de volta para o ER através de transporte retrógrado dependente dessa sequência KDEL para reconhecimento. O mecanismo é um pouco limpo – as proteínas que reconhecem e se ligam ao KDEL só o fazem a pH baixo, e o pH do Golgi é inferior ao do ER, por isso ligam o KDEL no Golgi, depois libertam-no quando estão de volta ao pH mais neutro do ER.
- Além disso, pense nisso, todas as proteínas que participam no transporte anterógrado – o V-SNARES, Rab, etc. – têm que voltar ao ER para que possam fazer tudo de novo, como o autocarro tem que voltar ao armazém do autocarro no final do dia.
- Como veremos em breve, o Golgi vem em múltiplas fases que dependem da adição de enzimas de mais a jusante.
O processo de transporte retrógrado não é tão diferente do anterógrado. Ele usa ARF ao invés de Sar1, COPI ao invés de COPII, mas funciona da mesma forma: ARF carregada com GTP deixa sua cauda hidrofóbica grudar na membrana, atraindo a atenção dos COPIs. O COPI tem dois componentes, COPIalpha e COPIbeta, ambos interagem com essa sequência KKXXX para recrutar proteínas ligadas à membrana destinadas ao transporte retrógrado. Algumas proteínas também têm uma seqüência RR (em qualquer parte da proteína) que pode sinalizá-las para transporte retrógrado.
O aparelho Golgi não é contíguo. É um conjunto empilhado de subcompartimentos separados chamados sacos ou cisternas. Os diferentes compartimentos têm propriedades e proteínas diferentes que os visitam numa ordem particular. Em ordem de ER para membrana celular, os compartimentos Golgi são chamados de cis, medial, trans e rede trans-Golgi. Cada compartimento tem diferentes enzimas que modificam as proteínas, e as modificações têm que acontecer em uma determinada ordem, daí a necessidade de um conjunto empilhado de compartimentos.
Mas como as proteínas amadurecem no Golgi, não é como se elas brotassem em vesículas de um compartimento e se movessem para o próximo. Ao invés disso, o compartimento em que elas já estão se move para fora e ‘amadurece’ à medida que novas enzimas são adicionadas a ele (de mais abaixo na cadeia do Golgi) via transporte retrógrado. Estranho, não é? É como se, em vez de mudar de uma escola primária para uma escola média para uma escola secundária, você apenas ficasse em um prédio escolar para toda a sua infância e adolescência, e eles apenas trouxessem novos livros didáticos e professores todos os anos para mantê-los adequados à nota que você e seus colegas de classe tinham agora alcançado. Aqui está como os Golgi são à medida que se movem e evoluem:
Então não há (pouco ou) transporte anterógrado dentro do Golgi, mas muito transporte retrógrado para trazer cada nova rodada de enzimas para dentro. Quando as proteínas finalmente completam o currículo completo do K-12 da rede Golgi, elas passam a ser transportadas para o seu destino final. Elas brotam em uma vesícula que irá para um dos três lugares:
- Exocitose – fusão com a membrana celular. Assim as proteínas lumenais serão secretadas extracelularmente, e as proteínas da membrana se tornarão proteínas da membrana celular.
- Vesículas secretas – estas apenas ficam como vesículas na célula até serem necessárias – onde ‘necessárias’ significa que elas eventualmente sofrem exocitose. Nos neurônios, é onde os neurotransmissores são armazenados até que um potencial de ação exija sua secreção para a sinapse. No estômago, as células que produzem enzimas gástricas mantêm essas enzimas em vesículas secretas até que a ingestão de alimentos provoque sua liberação no estômago.
- Lisossomos – onde proteínas desdobradas vão se degradar.
O transporte da rede trans-Golgi para esses destinos é diferente dos outros transportes discutidos acima e freqüentemente envolve clathrin (genes CLT__). As vesículas que brotam têm uma camada de duas camadas, com complexos proteicos adaptáveis (AP) como camada interna e a clathrina como camada externa. As proteínas adaptadoras têm um sinal alvo com um motivo YXXh (h = Φ = qualquer aminoácido hidrófobo). A clatrina forma a chamada formação ‘clathrin-triskelion’ mostrada aqui:

(Imagem graças ao usuário do Wikimedia Commons Phoebus87)
Clathrin também é responsável pela endocitose – o florescimento de vesículas de material extracelular (e proteínas da membrana celular) para entrar na célula. Isto é chamado de endocitose mediada por clathrina. Receptores na membrana celular são endocitosados com muita freqüência: toda a população de receptores hormonais gira a cada hora, especialmente quando os hormônios estão sendo recebidos. Levar o receptor para uma vesícula é uma maneira da célula cortar o sinal de entrada até que possa ser processado.
As notas sobre a membrana plasmática discutem brevemente a fibrose cística: CFTR é um transportador ABC responsável por bombear Cl- para fora da célula (ele também deixa Na+ entrar). Os mutantes com perda de função não bombeiam Cl-, o que remove a força motriz da osmose, engrossando o muco e causando problemas respiratórios. Existem pelo menos 127 mutantes CFTR diferentes com perda de função (pelo menos, é para isso que servem os testes Natera) que (se ambos os alelos estiverem desabilitados) causam fibrose cística. A mutação mais comum é ΔF508, que é ~3% de todos os alelos CFTR europeus e cerca de 70% dos alelos mutantes. A perda dessa fenilalanina altera a conformação do CFTR para que o código de saída diácida (aminoácidos D565 e D567) que visa o CFTR para vesículas exocitóticas não seja mais corretamente exposto e a proteína nunca chegue à membrana celular .
seção de discussão
seção que lemos Hu 2009, que mostrou que as proteínas de atlastin estão envolvidas na criação da rede de ER tubular. As evidências vieram quase inteiramente das interações proteína-proteína. Fiquei surpreendido por este artigo ter sido um grande negócio, porque houve um milhão de artigos mostrando interacções proteína-proteína para a Huntingtin, e ninguém acredita realmente em todas elas e isso não nos aproximou necessariamente de saber o que a Huntingtin faz ou o que corre mal na Doença de Huntington. Mas aparentemente Hu foi capaz de fazer um caso bastante limpo para as interacções dos atlastins com os reticulons como implicando um papel na formação de ER. Isso ajuda que Hu foi capaz de mostrar uma “interação genética” além de uma interação física (ligação). Uma ‘interação genética’ (eu tive que procurar) significa quando “às vezes mutações em dois genes produzem um fenótipo que é surpreendente à luz dos efeitos individuais de cada mutação”. Este fenômeno, que define a interação genética, pode revelar relações funcionais entre genes e caminhos”. .
PrP
Esta é uma década, então algumas coisas podem estar ultrapassadas, mas eu achei a revisão de Harris 2003 (ft) da biologia celular da PrP extremamente clara e útil. Kim & Hegde 2002 também foi de grande ajuda. PrP é uma proteína do caminho secreto. Os seus primeiros 22 aminoácidos (MANLGCWMLVLFVATWSDLGLC) são um peptídeo de sinal que causa a translocação cotradicional para as ER. Normalmente, o PrP apenas se liga ao seu terminal C e é ancorado ao lado exoplásmico da membrana. Mas os aminoácidos 111-134 (HMAGAAAAGAVVGGLGGYMLGSAM) são uma espécie de fraca sequência de ancoragem do sinal (Tipo II, com os +++ aminoácidos que vêm antes da ancoragem do sinal) que às vezes mas nem sempre se torna um domínio transmembrana, invertendo o terminal C no lúmen. Ainda mais confuso, essa sequência pode por vezes acabar como um domínio transmembrana sem a inversão, de modo que o terminal N está no lúmen. Assim, existem três topologias de membrana do PrP: a antiga e regular GPI-anchored, e duas orientações transmembrana, como descrito em Harris 2003 Fig 3:
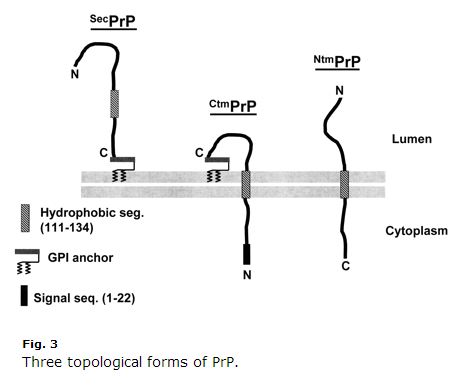
Note como o CtmPrP é esquisito. É transmembrana, mas também ancorada em GPI, e o peptídeo de sinal N-terminal nunca é clivado. Normalmente, as formas transmembrana são < 10% do total do PrP. Em algumas condições de laboratório a porcentagem é maior, e duas das mutações causadoras de GSS (A117V e P105L) também aumentam a fração de CtmPrP para 20-30% de toda a PrP. Destas três formas, há uma boa quantidade de evidências de que a CtmPrP é tóxica, e que pode desempenhar um papel na formação do prião, embora a maioria das mutações genéticas da doença do prião (incluindo a FFI D178N) não pareça afetar a topologia da membrana da PrP ou a fração da CtmPrP.
Após a PrP passar pelo Golgi, ela é direcionada para a membrana celular. Mas de acordo com Harris, ele não se limita a sentar ali – frequentemente através da endocitose mediada por clathrin e cicla através da célula a cada ~60 minutos, com algumas moléculas sendo clivadas em cada ciclo. O cobre estimula esta endocitose da PrP. A maioria das mutações da doença priônica genética muda a localização da PrP – geralmente quando uma mutação está presente, menos PrP é encontrada na superfície da célula, com mais acumulações na ER.