WPROWADZENIE
Amyloidoza jest chorobą depozycyjną spowodowaną pozakomórkowym gromadzeniem się fibryli, których źródłem są białka o niestabilnej strukturze, które ulegają fałdowaniu, agregacji i odkładaniu.1 Takie odkładanie może zmieniać strukturę tkanek i upośledzać funkcje różnych narządów i układów.2
Fibryle amyloidowe są nierozpuszczalne i odporne na proteolizę i są zwykle barwione czerwienią Kongo, wykazując intensywną żółto-zieloną dwójłomność w świetle spolaryzowanym.3 Ponad 30 białek może powodować odkładanie amyloidu, ale tylko 5 powoduje znaczne odkładanie w tkance sercowej1:
- –
Łańcuchy lekkie, które powodują amyloidozę pierwotną (AL).
- –
Transtyretyna (TTR), która powoduje amyloidozę TTR (ATTR).
- –
Apolipoproteina A.
- –
Fibrynogen.
- –
Białko amyloidowe A w surowicy, które wywołuje amyloidozę wtórną.
Amyloidoza pierwotna i ATTR są najczęstszymi postaciami amyloidozy sercowej, przy czym postać AL jest historycznie uważana za częstszą w krajach rozwiniętych.3.
Większość informacji na temat amyloidozy sercowej opiera się na AL. Jednak, chociaż liczba pacjentów z AL pozostaje stabilna, liczba rozpoznań ATTR ostatnio wzrosła i obecnie uważa się, że ATTR może występować znacznie częściej niż AL.2
Amyloidoza transtyretynowa bardzo często była przedmiotem błędnej diagnozy lub znacznych opóźnień w jej prawidłowym rozpoznaniu. Przyczyny tego stanu rzeczy to heterogenność jej postaci, konieczność potwierdzenia histologicznego, brak specjalistycznego sprzętu oraz błędne przekonanie niektórych kardiologów, że jest to choroba rzadka, bez możliwości leczenia.2,3
Ale te aspekty ulegają zmianie. Rozpoznanie ma wpływ na postępowanie z pacjentem. Opracowano specyficzne terapie, które mogą opóźnić lub ustabilizować odkładanie się złogów i które są bardziej skuteczne we wczesnych stadiach. Wczesna diagnoza jest zatem kluczowa. W niniejszym przeglądzie opisano istotne ostatnie postępy w diagnostyce i leczeniu ATTR, dając nadzieję pacjentom z tym schorzeniem.
TRANSTYRETYNA CARDIAC AMYLOIDOSIS
Transtyretyna jest tetramerycznym białkiem osocza odpowiedzialnym za transport tyroksyny i białek związanych z retinolem. Jest syntetyzowana głównie w wątrobie, a wtórnie w splocie naczyniówkowym i nabłonku barwnikowym siatkówki.4
Transtyretyna ma tendencję do dysocjacji do dimerów i monomerów, które błędnie łączą się w fibryle i ulegają odkładaniu. Mutacje punktowe lub wpływ wieku mogą zwiększyć tę tendencję, dając początek 2 klinicznym postaciom ATTR: zmutowanej (ATTRm) i typu dzikiego (ATTRwt).
MUTANTNA AMYLOIDOZA TRANSTYRETYNY
Obecnie wiadomo, że ponad 120 mutacji powoduje ATTRm. Mutacje te wykazują autosomalny dominujący sposób dziedziczenia, ze zmienną penetracją.4 Ze względu na duże zróżnicowanie geograficzne trudno jest ustalić częstość występowania ATTR, ale uważa się, że jest to choroba rzadka, z częstością występowania poniżej 1/100 000 mieszkańców2 (Tabela 1).
Główna charakterystyka kliniczna i diagnostyczna zmutowanej i dzikiej transtyretyny sercowej.Type Transthyretin Cardiac Amyloidosis
| ATTRwt | ATTRm | |
|---|---|---|
| Częstość występowania | Nieznana. Najwyraźniej bardzo częsta | |
| Badanie genetyczne | Brak mutacji w TTR | Mutacja w TTR |
| Typowy wiek w momencie wystąpienia | > 60 lat | Zmienny w zależności od mutacji sprawczej |
| Płeć | Przewaga mężczyzn. 80% pacjentów | Przewaga mężczyzn, z bardziej agresywnym fenotypem |
| Objawy pozasercowe | – Zespół cieśni nadgarstka (33%-49%) – Zwężenie odcinka lędźwiowego kręgosłupa – Urazowe zerwanie ścięgna mięśnia dwugłowego ramienia (32%) |
– Wstępująca obustronna polineuropatia czuciowo-ruchowa – Dysautonomia: niedociśnienie ortostatyczne, biegunka – zaparcia, zaburzenia erekcji – Zajęcie oka: jaskra, depozycja wewnątrzgałkowa, łuskowate źrenice |
| Upośledzenie czynności serca | Stałe | Zmienne w zależności od mutacji sprawczej |
| Wydolność serca | – Niewydolność serca (53%-86%) – Zaburzenia przewodzenia – Migotanie przedsionków (43%-67%) – Zwyrodnieniowy AoS |
– Zaburzenia przewodzenia – Niewydolność serca – Nierzadkie AF (10%) |
| Techniki diagnostyczne | ||
| EKG | – Wzorzec pseudoinfarktu (63%-66%) – Niskie napięcie (22%-33%) – Sokołowski LVH (6%-13%) |
– Pseudoinfarct pattern (18%-69%) – Niskie napięcie (2%-25%) – Sokołowski LVH (3%-8%) |
| ECHO | – Moderate-severe hypertrophy – Mild-moderate depressed LVEF (30%) |
– Moderate hypertrophy – LVEF, typowo zachowana |
| MRI serca | – Późne wzmocnienie – Podwyższone natywne T1 i EV |
|
| Scyntygrafia 99mTc DPD | – Stopień 2-3 | – Stopień 0: bezobjawowi nosiciele – Stopień 1: początkowe zajęcie serca – Stopień 2-3: znaczne zaangażowanie serca |
AF, migotanie przedsionków; AoS, stenoza aortalna; ATTRm, zmutowana amyloidoza transtyretyny; ATTRwt, amyloidoza transtyretyny typu dzikiego; ECG, elektrokardiogram; ECO, echokardiogram; EV, objętość pozakomórkowa; LVEF, frakcja wyrzutowa lewej komory; LVH, przerost lewej komory; TTR, transtyretyna.
Pierwsze mutacje TTR odnotowano jako rodzinną polineuropatię amyloidową (lub chorobę Andrade), w związku z czym ATTRm do niedawna uważano za chorobę neurologiczną. Jednak ostatnie wyniki badań wskazują na zajęcie serca w ponad połowie przypadków.3
Istnieje silna korelacja genotyp-fenotyp, przy czym mutacje są związane z chorobą czysto neurologiczną lub czysto kardiologiczną.3 Jednakże podział ATTRm na chorobę kardiologiczną lub neurologiczną może być zbytnim uproszczeniem, ponieważ te dwie postacie kliniczne w spektrum choroby w znacznym stopniu się pokrywają.3
Mutacja Val30Met (obecnie znana jako Val50Met po dodaniu 20 pozycji do tradycyjnej nazwy mutacji w ATTRm) jest najczęstszą mutacją na świecie i występuje endemicznie w Portugalii, Japonii i Szwecji. Szacowana częstość jej występowania w Portugalii wynosi 1 na 538 mieszkańców.2 Majorka (Hiszpania) i Valverde del Camino (Huelva, Hiszpania) są również uważane za obszary, w których ATTRm występuje endemicznie. Szacowana częstość występowania na Majorce u pacjentów z objawami wynosi 3/100 000 mieszkańców.5
Mutacja Val30Met powoduje głównie schorzenie neurologiczne z symetryczną polineuropatią czuciowo-ruchową, która rozpoczyna się w kończynach dolnych i ma charakter wstępujący. Może być związana z dysautonomią z niedociśnieniem ortostatycznym, zaburzeniami erekcji, nietrzymaniem moczu i objawami żołądkowo-jelitowymi. Zwykle rozpoczyna się pod koniec drugiej lub w trzeciej dekadzie życia, a u 43% pacjentów dochodzi do zajęcia serca, co jest częstą przyczyną zgonu4 (tab. 1).
Szczególnie istotna jest mutacja Val122Ile (p. Val142Ile), która występuje u 3% do 4% północnoamerykańskiej populacji rasy czarnej.3 Chociaż jej penetracja jest niekompletna,3 mutacja ta wiąże się z 47% zwiększonym ryzykiem rozwoju niewydolności serca (HF).6 Ostatnie badanie wykazało, że amyloidoza Val122Ile była czwartą najczęstszą przyczyną HF w brytyjskiej populacji afro-karaibskiej.7 Chociaż do 30% pacjentów z tą mutacją może mieć cechy łagodnej neuropatii,6 fenotyp kliniczny jest zwykle podobny do fenotypu ATTRwt.4 Val122Ile nie powinien być uważany za mutację wyłącznie dla populacji czarnej, ponieważ może być również obecny w populacji białej. Na przykład, zidentyfikowaliśmy tę mutację w 4 białych hiszpańskich rodzinach bez czarnych przodków.
WILD-TYPE TRANSTHYRETIN AMYLOIDOSIS
Amyloidoza transtyretynowa typu Wilda została po raz pierwszy opisana w 1876 roku. Dawniej nazywano ją amyloidozą starczą, ale jej rozpoznanie u pacjentów w wieku od 40 do 60 lat spowodowało, że termin ten stał się nieaktualny. Co ciekawe, najwcześniejszy znany przypadek tej mutacji został znaleziony u 47-letniego amerykańskiego pacjenta.8
Dokładna częstość występowania ATTRwt pozostaje nieznana. Badania sugerują jednak, że jest ona niedostatecznie diagnozowana i że może być najczęstszą postacią amyloidozy sercowej.2,3 Hipotezę tę potwierdzają następujące wyniki:
- –
U pacjentów w wieku powyżej 80 lat częstość występowania odkładania się TTR wynosi 25% w badaniu autopsyjnym.3
- –
W przypadku pacjentów z HF z zachowaną frakcją wyrzutową (HFpEF) umiarkowanie-ciężkie odkładanie TTR wynosi 5% w badaniu autopsyjnym.9
- –
Wśród pacjentów w wieku ponad 60 lat przyjmowanych z powodu HFpEF i przerostu lewej komory (LVH) ≥ 12 mm nasza grupa stwierdziła ostatnio częstość występowania choroby na poziomie 13%.10
W przeciwieństwie do ATTRm, ATTRwt jest chorobą występującą sporadycznie, której początek typowo przypada po 70. roku życia.4 Występuje głównie u mężczyzn, a opublikowane serie donoszą o częstości występowania od 89% do 98%.11,12 Jednak w ostatniej serii pacjentów, u których zdiagnozowano ATTRwt w 2 szpitalach (Madryt, Hiszpania i Bolonia, Włochy), nasza grupa stwierdziła, że 20% stanowiły kobiety. Co więcej, inne badania autopsyjne również sugerują, że ATTRwt u kobiet może być bardziej rozpowszechniona niż wcześniej donoszono. Dlatego płeć żeńska nie powinna zmniejszać klinicznego podejrzenia ATTRwt (Tabela 1).13
Wyniki autopsji wskazują, że w ATTRwt odkładanie TTR jest rozproszone w różnych narządach. Jednak ze względu na tropizm sercowy TTR odkładanie jest znacznie większe w sercu, a zajęcie serca jest główną manifestacją kliniczną.4 U pacjentów mogą występować objawy pozasercowego odkładania TTR, takie jak zwężenie kanału lędźwiowego, atraumatyczne zerwanie ścięgna mięśnia dwugłowego lub „objaw Popeye’a” oraz zespół cieśni nadgarstka (CTS)3 (ryc. 1). Wszystkie te cechy mogą pomóc w postawieniu diagnozy i szybkim jej ustaleniu. CTS może towarzyszyć innym podtypom amyloidozy, ale jest częstszy w ATTRwt. Depozycja może poprzedzać objawy sercowe o kilka lat.6 Może być wskazaniem u starszych pacjentów z LVH, zwłaszcza jeśli mają obustronny CTS niezwiązany z konkretnymi czynnościami zawodowymi i znajdują się w klasie czynnościowej ≥ II według New York Heart Association (dane niepublikowane).
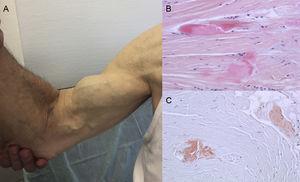
Znaki i objawy amyloidozy transtyretynowej. A: nieurazowe zerwanie ścięgna prawego bicepsa („objaw Popeye’a”). B i C: barwienie hematoksyliną-eozyną (B) i czerwienią Kongo (C), oba ×200, próbki więzadła nadgarstka pokazujące gęste wiązki kolagenowe z materiałem niekomórkowym. Dzięki uprzejmości dr Clary Salas Antón.
DIAGNOZA AMYLOIDOZY TRANSTYRETYNYPrezentacja kliniczna
Amyloid może naciekać każdą strukturę serca.1 Zazwyczaj odkładanie się amyloidu zwiększa grubość ścian komór, co powoduje stopniowe zmniejszanie się ich rozciągliwości, prowadząc do poważnej dysfunkcji rozkurczowej. Dlatego też ATTR tradycyjnie zalicza się do przyczyn kardiomiopatii restrykcyjnej.
Jednakże spektrum kliniczne ATTR jest znacznie szersze i bardziej heterogenne. Najczęstszym objawem ATTR jest HF. Jak wspomniano, w badaniu opublikowanym przez naszą grupę w 2015 r. zasugerowano, że protokół oparty na scyntygrafii z użyciem kwasu 99mTc-3,3-difosfono-1,2-propanodikarboksylowego (99mTc-DPD) może być przydatny w rozpoznawaniu ATTRwt u znacznego odsetka (13%) pacjentów w wieku powyżej 60 lat przyjętych z powodu HFpEF.10 Na podstawie tego wyniku scyntygrafię z użyciem 99mTc-DPD włączono do europejskich wytycznych dotyczących HF z 2016 r. jako przydatne narzędzie do identyfikacji pacjentów z ATTR.14. Nie należy jednak podejrzewać ATTR wyłącznie u pacjentów z HFpEF, ponieważ wraz z postępem odkładania amyloidu pogarsza się funkcja skurczowa, a w konsekwencji ATTR może być związana z różnym stopniem dysfunkcji skurczowej.
Amyloidoza transtyretyny jest fenokopią kardiomiopatii przerostowej (HCM) i może być z nią mylona. Ostatnie wieloośrodkowe badanie francuskie wykazało, że 5% pacjentów z HCM ma ATTRm.15 Jednak nasze wyniki nie są zgodne z tym wysokim wskaźnikiem, co może być związane z dużą populacją osób rasy czarnej we Francji.
Zaburzenia przewodzenia w sercu mogą być pierwszą manifestacją ATTR. Naciekanie amyloidem węzłów zatokowych i przedsionkowo-komorowych1 może wskazywać na konieczność wszczepienia stymulatora serca (tab. 1). We wspomnianym wcześniej badaniu przeprowadzonym w Hiszpanii i we Włoszech stwierdzono, że zaburzenia przewodzenia były pierwszą manifestacją ATTRwt u 7% pacjentów z tą chorobą.13
Arytmie przedsionkowe są również bardzo częste u pacjentów z ATTRwt13 (ryc. 2A), a pierwszą manifestacją choroby może być udar mózgu.4 W rzeczywistości grupa Mayo Clinic zasugerowała ostatnio, że ATTRwt należy wykluczyć w przypadku rozpoznania niezastawkowego migotania przedsionków (AF) u pacjentów w podeszłym wieku.8 Z kolei AF występuje znacznie rzadziej u pacjentów z ATTRm (Tabela 1).
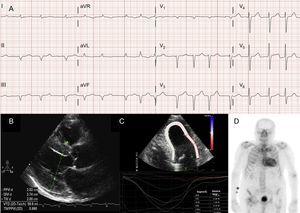
Techniki diagnostyczne w amyloidozie transtyretynowej serca (ATTR). A: elektrokardiogram pacjenta z amyloidozą transtyretynową typu dzikiego (ATTRwt), ukazujący migotanie przedsionków i wzór pseudoinfarktu w odprowadzeniach dolnych. B: echokardiogram pacjenta ze zmutowaną amyloidozą transtyretynową z mutacją Val30Met, z zaznaczonym koncentrycznym przerostem lewej komory i łagodnym wysiękiem osierdziowym. C: Podłużne odkształcenie regionalne u pacjenta z ATTRwt, wykazujące zachowane wartości w segmencie koniuszkowym i obniżone wartości w segmencie podstawnym i środkowo-komorowym. D, skan 99mTc-DPD (kwas 99mTc-3,3-difosfono-1,2-propanodikarboksylowy) u pacjenta z ATTRwt, wykazujący wychwyt obojczykowy przewyższający wychwyt kostny, odpowiadający stopniowi 3 w skali Peruginiego.
Wreszcie, zauważamy, że ATTR i zwyrodnieniowa stenoza aortalna mogą współistnieć u tego samego pacjenta. W 2016 roku w kilku badaniach zwrócono uwagę na taką możliwość, a w badaniu prospektywnym odnotowano, że częstość występowania ATTRwt wynosiła 6% u pacjentów w wieku powyżej 65 lat, u których przeprowadzono wymianę zastawki aortalnej.16 Badanie to sugerowało, że pacjenci z obiema jednostkami mieli znacznie gorsze rokowanie pooperacyjne niż pacjenci bez ATTRwt (śmiertelność 50% vs 6,9% po medianie obserwacji wynoszącej 2,3 roku).16 W innym niedawnym badaniu z zastosowaniem scyntygrafii 99mTc-DPD u 43 pacjentów ze zwężeniem aortalnym o małym przepływie/niskim gradiencie zidentyfikowano 5 pacjentów z ATTRwt (częstość występowania 12%). 17 Pacjenci z ciężką stenozą aortalną i ATTRwt mają ten sam profil demograficzny, a odpowiednie leczenie pacjentów z obiema chorobami wciąż nie zostało określone.
Użyteczność technik diagnostycznych
Rozpoznanie ATTR stanowi wyzwanie w codziennej praktyce klinicznej. Chociaż elektrokardiografia i echokardiografia odgrywają pewną rolę w diagnostyce, nowe techniki nieinwazyjne zyskały kluczową rolę w ocenie pacjentów z ATTR.
Elektrokardiogram
Związek między niskim napięciem a amyloidozą serca od dawna uważany jest za niepodważalny.3 Najczęściej stosowanymi kryteriami w praktyce klinicznej są amplituda QRS.1 Chociaż niskie napięcie elektrokardiograficzne w otoczeniu LVH powinno wzbudzić podejrzenie, częstość występowania we współczesnej serii ATTR wynosiła od 20% do 25%.3,4,13 Częstość występowania różni się również w zależności od stosowanych kryteriów. Na przykład zastosowanie kryterium Sokołowa (załamek S w odprowadzeniu V1 + załamek R w odprowadzeniu V5 lub V6
1,5 mV) może zwiększyć obliczoną częstość występowania do 46%-58%.13 Zaleca się stosowanie stosunku grubości ściany lewej komory do całkowitego napięcia QRS w celu lepszej oceny rozbieżności między wynikami obu technik.2,3 Jednak nawet 20% pacjentów z ATTR może spełniać elektrokardiograficzne kryteria LVH.2,3
W większości serii pacjentów z amyloidozą serca najczęstszym objawem elektrokardiograficznym jest pseudoinfarktacja2,3,13 (ryc. 2A). Ze względu na możliwe zajęcie układu przewodzącego, częste są również całkowite lub niekompletne bloki odnóg pęczka Hisa.3
Echokardiografia
Chociaż echokardiografia jest podstawą wstępnej diagnostyki ATTR, żadne wyniki nie są specyficzne.3 Amyloidoza transtyretyny jest zwykle związana z prawidłową lub małą lewą komorą z koncentrycznym przerostem.3 Podczas 10. międzynarodowego sympozjum na temat amyloidu i amyloidozy, które odbyło się w 2004 r., ustanowiono echokardiograficzne kryterium choroby serca spowodowanej AL przy braku innych przyczyn LVH jako obecność LVH z wartością odcięcia 12 mm dla grubości ściany przegrody międzykomorowej.4 Kryterium to zostało później ekstrapolowane na inne formy amyloidozy (ryc. 2B), co zapewniło wysoki stopień swoistości, ale niską czułość.
Chociaż klasycznie opisywano koncentryczny LVH, aktualne serie sugerują, że około 20% ma asymetryczny LVH.13
Mimo klasycznego związku między prawidłową lub nieznacznie obniżoną frakcją wyrzutową lewej komory (LVEF) a amyloidozą serca,2 zakres LVEF jest bardzo zmienny.8 W ostatnim badaniu przeprowadzonym w Mayo Clinic, LVEF 8, podczas gdy w naszej serii LVEF 13 Ponadto, wykorzystanie LVEF w ocenie funkcji skurczowej w amyloidozie serca jest ograniczone, ponieważ nieznacznie obniżone wartości są już wskaźnikiem istotnej choroby serca. Ograniczenie to można przezwyciężyć, stosując prędkości doplera tkankowego, obrazowanie odkształcenia i frakcję skurczu mięśnia sercowego, które zaproponowano jako bardziej odpowiednie wskaźniki oceny funkcji serca.2
Inne klasyczne objawy echokardiograficzne to przerost prawej komory, rozstrzeń dwudzielna, łagodny wysięk w osierdziu, pogrubienie zastawki przedsionkowo-komorowej, pogrubienie ściany przegrody międzyprzedsionkowej i ziarnisty, lśniący wygląd mięśnia sercowego.3,6 Ponieważ jednak niektóre z tych cech obserwowano w wysoce wyselekcjonowanej serii pacjentów w zaawansowanym stadium choroby, nie wszystkie z nich muszą być obecne, aby wzbudzić podejrzenie.1
Obrazy odkształcenia regionalnego są bardzo przydatną techniką we wczesnej diagnostyce pacjentów z ATTR. U pacjentów z ATTR odkształcenie podłużne jest obniżone w segmentach podstawnych i środkowo-komorowych, ale jest zachowane w segmentach koniuszkowych18 (ryc. 2C). Ten typowy wzorzec może być przydatny w diagnostyce różnicowej ATTR z innymi chorobami serca.4
Biomarkery
Jest mniej danych na temat roli N-końcowego prohormonu mózgowego propeptydu natriuretycznego (NT-proBNP) i troponiny w ATTR niż w AL.4 Poziomy NT-proBNP w ATTR są zwykle niższe niż w AL,4 co odzwierciedla 2 różne mechanizmy patofizjologiczne: bezpośrednią toksyczność łańcucha lekkiego w AL vs indukowane uszkodzenie tkanki przez protofibryle w ATTR.
Ostatnio grupa Mayo Clinic zaproponowała system stratyfikacji podobny do tego, który obowiązuje w przypadku AL. W kohorcie 360 pacjentów z ATTRwt wykazano, że oba biomarkery są predyktorami śmiertelności. Pacjenci w stadium III (NT-proBNP > 3000 pg/mL i troponina T > 0,05 ng/mL) mieli medianę przeżycia wynoszącą 20 miesięcy, podczas gdy pacjenci w stadium I i II mieli medianę przeżycia wynoszącą 66 miesięcy i 40 miesięcy (odpowiednio brak biomarkera lub tylko 1 biomarker powyżej ustalonych punktów odcięcia).
Rezonans magnetyczny serca
Rezonans magnetyczny serca (CMRI) może być stosowany w celu uzyskania informacji strukturalnych i czynnościowych oraz scharakteryzowania składu tkanki mięśnia sercowego.3 Badanie CMRI jest niezbędne we wczesnej identyfikacji ATTR oraz w jego diagnostyce różnicowej z innymi chorobami serca.
Charakterystyka tkanki za pomocą CMRI opiera się na następujących cechach:
- –
Późne wzmocnienie: Globalny wzór podwsierdziowy jest praktycznie patognomoniczny dla amyloidozy serca, ale jest obecny tylko u około jednej czwartej pacjentów. Inne wzorce, takie jak transmuralny (najczęstszy) lub łatanie, są również zgodne (rysunek 3). Pomimo wysokiej czułości i swoistości należy wziąć pod uwagę możliwość braku późnego wzmocnienia (15% pacjentów) oraz, jak wynika z naszego doświadczenia, nieistotny odsetek wyników fałszywie ujemnych z przyczyn technicznych.3 Wzorzec transmuralnego wzmocnienia wiąże się z gorszym rokowaniem i jest niezależnym predyktorem śmiertelności.19
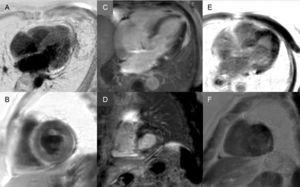 Rycina 3.
Rycina 3.Zróżnicowanie wzorów późnego wzmocnienia w rezonansie magnetycznym serca w amyloidozie transtyretynowej. A i B: sekwencje późnego wzmocnienia, odpowiednio płaszczyzna 4-jamowa i oś krótka na poziomie środkowym, u pacjenta ze zmutowaną amyloidozą transtyretynową (ATTRm), wykazujące rozproszone patologiczne transmuralne odkładanie gadolinu. C i D: sekwencje późnego wzmocnienia, odpowiednio 4-jamowa i krótkoosiowa na poziomie podstawnym, u pacjenta z ATTRm, wykazujące patologiczne odkładanie gadolinu o wzorze plamistym, z dolnym inferoseptalnym i inferolateralnym ogniskiem podstawnym. E i F, późne sekwencje wzmocnienia, płaszczyzna 4-jamowa i oś krótka na poziomie koniuszka, odpowiednio, u pacjentów z ATTRm, pokazujące rozległe patologiczne transmuralne odkładanie, z wyjątkiem segmentów podstawnych i środkowych przednio-bocznych. Dzięki uprzejmości Dr Jesús González Mirelis.
(0.15MB). - –
Długie czasy T1: Mapowanie T1 jest techniką, w której ilościowy sygnał mięśnia sercowego jest mierzony przed (natywny T1) lub po podaniu kontrastu. Natywny czas T1 jest bardzo długi w amyloidozie serca.3 Mapowanie T1 nie wymaga podawania kontrastu i dlatego może być stosowane w niewydolności nerek. Czasy T1 mogą być nieprawidłowe nawet przed zaobserwowaniem LVH.3 Czasy T1 są dłuższe w ATTR niż w HCM i grupie kontrolnej (1097 ms ± 43 ms vs 1026 ms ± 64 ms vs 9,67 ms ± 34 ms, odpowiednio; P
ms ± 68 ms; P = 0,01).20
Podanie kontrastu można wykorzystać do obliczenia objętości pozakomórkowej (extracellular volume, ECV) i oceny zwiększenia przestrzeni pozakomórkowej. Wartości ECV w amyloidozie sercowej są wyższe niż w innych chorobach serca, z wyjątkiem stref zawału mięśnia sercowego.21 W 2016 r. nasza grupa, we współpracy z innymi ośrodkami krajowymi, doniosła, że kwantyfikacja ECV może zidentyfikować zajęcie serca w ATTRm i po raz pierwszy skorelowała ją ze stopniem upośledzenia neurologicznego, wspierając zastosowanie tej techniki we wczesnej diagnostyce i śledzeniu ATTRm.22
Ilościowe techniki mapowania T1 i obliczania ECV mogą być bardzo przydatne w ATTR do wczesnej diagnostyki, obserwacji klinicznej i oceny odpowiedzi na leczenie (ryc. 4).
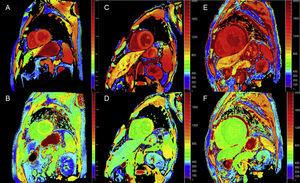
Mapowanie T1, przed i po podaniu kontrastu, ze zmodyfikowaną metodą look-locker inversion-recovery (MOLLI) w 3T rezonansie magnetycznym serca u zdrowej kontroli, pacjenta z amyloidozą transtyretynową i pacjenta z pierwotną amyloidozą łańcuchów lekkich. A i B: natywne mapowanie T1 i objętość zewnątrzkomórkowa (EV), odpowiednio, w zdrowej grupie kontrolnej, wykazujące wartości prawidłowe (EV = 0,214). C i D: natywne mapowanie T1 i EV, odpowiednio, u pacjenta ze zmutowaną amyloidozą transtyretynową z uszkodzeniem neurologicznym i początkiem zajęcia serca, podwyższonym natywnym T1 i nieznacznie podwyższonym EV (0,361). E i F: mapowanie natywnego T1 i EV, odpowiednio, u pacjenta z amyloidozą sercową transtyretyny typu dzikiego, podwyższone natywne T1 i bardzo wysokie EV (0,626), odzwierciedlające masywną infiltrację amyloidu. Dzięki uprzejmości dr Jesúsa Gonzáleza Mirelisa.
Scyntygrafia serca
W latach 80. ubiegłego wieku obserwacja wychwytu sercowego kilku znaczników bifosfonianów kostnych została histologicznie skorelowana z obecnością amyloidozy serca.23 Mechanizm wychwytu nie jest dobrze scharakteryzowany, ale może być związany z zawartością wapnia w złogach amyloidu.
Wczesne badanie przeprowadzone przez grupę bolońską z użyciem 99mTc-DPD wykazało wychwyt sercowy u 15 pacjentów z ATTR i jego brak u 10 pacjentów z AL, stosując punktację opartą na wychwycie obojczykowym równym lub wyższym od wychwytu kostnego (Perugini score)24 (ryc. 2D). Podobne wyniki zostały następnie przedstawione przez naszą grupę i innych.25 Łagodny wychwyt (wynik 1) i umiarkowany wychwyt (wynik 2) można znaleźć odpowiednio u 30% i 10% pacjentów z AL.24
Z uwagi na wysoką czułość i swoistość, technika ta jest niezwykle przydatna do ustalenia rozpoznania ATTR i może wykazać zajęcie serca, nawet jeśli wyniki echokardiografii i MRI są nadal prawidłowe. W rzeczywistości, po wykonaniu scyntygrafii ze wskazań onkologicznych lub reumatologicznych, przypadkowe wykrycie ATTR nie jest rzadkie.26
Tc-DPD nie jest dostępny w Stanach Zjednoczonych, ale podobne wyniki uzyskano stosując obrazowanie z użyciem 99mTc-PYP (pirofosforan).27
Inne radiotraktery są obecnie przedmiotem badań. Na przykład 18F-florbetapir, który został już zatwierdzony do obrazowania beta-amyloidu w mózgu,4 był badany u pacjentów z AL i ATTR. Wyniki wskazują, że 18F-florbetapir może wykrywać złogi AL i ATTR w mięśniu sercowym.28 Chociaż dostępne dane uzyskano w badaniach klinicznych29 , a wysoki koszt tego radioznacznika ogranicza jego zastosowanie, trwa kilka badań nad potencjalną przewagą jego zastosowania nad Tc-DPD jako techniki przesiewowej w 2 najczęstszych typach amyloidozy.
Diagnostyka inwazyjna
Ostateczne rozpoznanie ATTR opiera się na histologicznym wykazaniu obecności włókien amyloidu. Chociaż może występować odkładanie pozasercowe, prawdopodobieństwo wykazania amyloidu w badaniu histologicznym różni się w zależności od narządu.2 Istnieje niewiele badań dotyczących opłacalności biopsji pozasercowej (np. tłuszczu brzusznego, dziąseł, ślinianek, przewodu pokarmowego) w ATTR, która jest większa w ATTRm niż w ATTRwt. Jednak negatywny wynik biopsji narządu klinicznie niedotkniętego chorobą nie wyklucza rozpoznania ATTR.4
Tak jak w ATTRwt, biopsja endomiokardialna jest wskazana u pacjentów bez zajęcia narządów pozasercowych lub z samą chorobą serca.3,4 Biopsja endomiokardialna jest procedurą niskiego ryzyka (zwłaszcza w doświadczonych ośrodkach), a błędy w próbkowaniu są mało prawdopodobne.6
Po histologicznym potwierdzeniu amyloidozy, które czasami może wymagać interpretacji przez przeszkolony personel,6 kluczowe znaczenie ma prawidłowa klasyfikacja podtypu.4 Obecnie klasyfikacja zależy od połączenia immunohistochemii, analizy genetycznej i proteomiki:
- –
Immunohistochemia opiera się na zastosowaniu swoistych przeciwciał przeciwko znanym białkom amyloidowym. Chociaż wyniki tej techniki są zazwyczaj ostateczne, jest ona mniej czuła w rozpoznawaniu łańcuchów lekkich.4
- –
To ograniczenie można przezwyciężyć stosując spektrometrię mas, która dostarcza ostatecznych wyników i jest standardem kryterium w potwierdzaniu podtypu amyloidu.2 Chociaż technika ta jest dostępna tylko w specjalistycznych ośrodkach, jest ona szczególnie przydatna w przypadkach niejednoznacznych lub w przypadkach dodatnich dla kilku przeciwciał w badaniu immunohistochemicznym, co z naszego doświadczenia zdarza się w około 20% do 30% przypadków. 4
- –
Ponieważ techniki kliniczne i histologiczne nie są w stanie odróżnić ATTRm od ATTRwt, badania genetyczne są zalecane we wszystkich przypadkach ATTR. Znalezienie mutacji przyczynowej może mieć znaczenie przy oferowaniu poradnictwa genetycznego i obserwacji bezobjawowych nosicieli, 4,30 którzy mogą odnieść korzyści z nadchodzących terapii opóźniających wystąpienie choroby lub nawet zapobiegających jej wystąpieniu.31
Diagnostyka nieinwazyjna
Do niedawna badania histologiczne uważano za niezbędne w diagnostyce ATTR.3 Aby jednak ułatwić diagnostykę, w 2016 roku w międzynarodowym wieloośrodkowym badaniu zaproponowano nowy algorytm nieinwazyjnej diagnostyki ATTR.32
W badaniu tym przeanalizowano wyniki 1217 pacjentów. Obecność klasycznych objawów amyloidozy serca przy użyciu technik obrazowania, wychwyt Tc-DPD/PYP stopnia 2 lub 3 w scyntygrafii oraz brak białka monoklonalnego miały swoistość i dodatnią wartość predykcyjną dla ATTR wynoszącą 100%32 (ryc. 5).
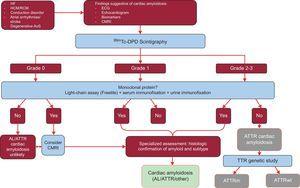
Algorytm diagnostyczny dla pacjentów z podejrzeniem amyloidozy serca. System klasyfikacji scyntygrafii 99mTc-DPD: stopień 0, brak wychwytu w sercu; stopień 1, nieznacznie niższy wychwyt niż w kości; stopień 2, umiarkowany wychwyt równy wychwytowi w kości; stopień 3, znaczny wychwyt przewyższający wychwyt w kości. ACV, udar; AL, pierwotna amyloidoza łańcuchów lekkich; AoS, stenoza aortalna; ATTR, amyloidoza transtyretyny; ATTRm, zmutowana amyloidoza transtyretyny; ATTRwt, amyloidoza transtyretyny typu dzikiego; CMRI, rezonans magnetyczny serca; ECG, elektrokardiogram; HCM, kardiomiopatia przerostowa; HF, niewydolność serca; RCM, kardiomiopatia restrykcyjna; TTR, transtyretyna.
Kluczową cechą tego algorytmu jest nieobecność białka monoklonalnego, które mogłoby powodować AL w surowiczym teście łańcuchowym (Freelite, The Binding Site, UK) oraz w elektroforezie immunofiksacyjnej krwi i moczu. Obecność białka monoklonalnego jest wskazaniem do wykonania biopsji endomiokardialnej w celu odróżnienia ATTR od AL.32 Do 5% populacji w wieku powyżej 65 lat ma gammopatię monoklonalną o nieustalonym znaczeniu.2 U osób w podeszłym wieku umiarkowany wzrost krążących łańcuchów lekkich nie powinien bezpośrednio prowadzić do rozpoznania AL. Donoszono, że do 10% pacjentów w podeszłym wieku z ATTRwt i gammopatią monoklonalną o nieustalonym znaczeniu w ośrodkach referencyjnych otrzymało wcześniej błędne rozpoznanie AL.3,33 Prawidłowe rozpoznanie jest konieczne, aby uniknąć niewłaściwej chemioterapii. Co ciekawe, w naszym szpitalu udokumentowano 2 przypadki pacjentów ze szpiczakiem mnogim i współistniejącym ATTRwt na podstawie spektrometrii mas.
LECZENIE AMYLOIDOZY KARDIACZA TRANSTYRETYNOWEGO
Leczenie pacjentów z ATTR ma 2 cele: zapewnienie wsparcia medycznego oraz, jeśli to możliwe, zatrzymanie lub opóźnienie odkładania amyloidu poprzez zastosowanie określonych metod leczenia.
Postępowanie medyczne
Następujące rozdziały opisują wspomagającą opiekę kardiologiczną nad pacjentami z ATTR.
Postępowanie w niewydolności serca
U pacjentów z amyloidozą sercową należy utrzymywać wolemię. Dieta i styl życia są bardzo ważne. Diuretyki mają kluczowe znaczenie w leczeniu HF w ATTR. Ponieważ jednak nadmierne stosowanie diuretyków może prowadzić do hipotensji (często z powodu dysfunkcji autonomicznej) i pogorszenia sytuacji klinicznej, zwłaszcza w ATTRm, należy zachować szczególną ostrożność w ich stosowaniu.
W leczeniu HF w ATTR należy wziąć pod uwagę, że upośledzona funkcja rozkurczowa i zmniejszona objętość wyrzutowa prowadzą do kompensacyjnej tachykardii w celu utrzymania rzutu serca. Dlatego beta-blokery muszą być stosowane ostrożnie i indywidualnie dla każdego pacjenta. Standardową praktyką jest ich odstawianie w przypadku braku trudności w kontroli częstości akcji serca. Takie podejście jest jeszcze ważniejsze, jeśli to możliwe, w ATTRwt ze względu na częste występowanie zaburzeń przewodzenia.6 Antagoniści wapnia i digoksyna mogą wiązać się z włóknami amyloidu i dlatego są przeciwwskazane w ATTR ze względu na ryzyko toksyczności nawet w dawkach terapeutycznych.6
W przeciwieństwie do HF z dysfunkcją skurczową spowodowaną innymi przyczynami, nie ma dowodów na korzyści prognostyczne wynikające ze stosowania beta-blokerów, inhibitorów konwertazy angiotensyny lub antagonistów receptora angiotensyny II w amyloidozie serca. W rzeczywistości ich stosowanie może prowadzić do pogorszenia stanu klinicznego z powodu hipotensji i niskiego rzutu serca: w jednej z ostatnich publikacji odnotowano gorsze rokowanie w ATTRm i neutralny efekt w ATTRwt.34
Zarządzanie arytmiami przedsionkowymi
Postępowanie z migotaniem przedsionków w ATTR stanowi wyzwanie. Utrzymanie długotrwałego rytmu zatokowego jest trudne. Można jednak podjąć próbę kardiowersji elektrycznej, ponieważ może ona prowadzić do poprawy stanu klinicznego.
Ryzyko zakrzepowo-zatorowe u pacjentów z ATTR jest bardzo wysokie. Ponadto przewlekły naciek amyloidowy może prowadzić do mechanicznej dysfunkcji przedsionków, co może być przyczyną powstawania skrzeplin w przedsionkach u niektórych pacjentów bez AF. Leczenie przeciwzakrzepowe w ATTR nie powinno być oparte na skali CHADS2-VASC i powinno być standardową terapią w AF. Zdarzenia krwotoczne występują rzadziej niż w AL, dlatego niektóre szpitale zalecają leczenie przeciwkrzepliwe u pacjentów z rytmem zatokowym, jeśli funkcja przedsionków jest słaba, co można stwierdzić na podstawie prędkości przepływu krwi w Dopplerze przez zastawkę. Chociaż nie ma badań porównawczych dotyczących skuteczności bezpośrednich doustnych antykoagulantów w porównaniu z antagonistami witaminy K, w naszym szpitalu wybranym pacjentom podawano bezpośrednie doustne antykoagulanty.
Rola urządzeń
Obecne zalecenia dotyczące wszczepiania stymulatorów serca są takie same w ATTR i w populacji ogólnej. Niektóre grupy opowiadają się jednak za profilaktyczną implantacją, zwłaszcza u pacjentów z ATTRm i zaburzeniami przewodzenia.35 Nie popieramy tej strategii prewencyjnej i nie stwierdziliśmy tak dużej częstości występowania zaburzeń przewodzenia, aby uzasadniała ona profilaktyczną implantację u pacjentów z ATTRm.
Rola stosowania wszczepialnych kardiowerterów-defibrylatorów (ICD) w ATTR nie jest dobrze ustalona. W niewielkiej serii implantacja ICD nie poprawiła znacząco przeżycia, chociaż miała odpowiedni efekt u wielu pacjentów w ciągu pierwszych 2 lat.36
Przeszczep serca
Przeszczep serca odgrywa niewielką rolę w ATTR, ponieważ ATTRm może obejmować różne narządy, a ATTRwt zwykle dotyka pacjentów w podeszłym wieku. Jednak brak zajęcia narządów pozasercowych u pacjentów z ATTRwt czyni ich dobrymi kandydatami do zabiegu. W literaturze można znaleźć przykłady udanych przeszczepów u pacjentów młodszych niż 70 lat z ATTRwt lub z ATTRm i dominującym zajęciem serca.4
Szczególne leczenie amyloidozy sercowej transtyretynowej
Obecnie nie ma zatwierdzonej terapii do specyficznego leczenia amyloidozy sercowej ATTR, chociaż przeszczepienie wątroby (TxH) samodzielnie lub w połączeniu z przeszczepieniem serca jest stosowane w ATTRm od lat 90. jako sposób na wyeliminowanie głównego źródła prekursorowego TTR.4
Głównymi ograniczeniami tej techniki są jednak: niedobór dawców, konieczność przewlekłej immunosupresji, zaawansowany wiek w momencie prezentacji oraz gorsze wyniki uzyskiwane u pacjentów z mutacjami innymi niż mutacja Val30Met.
Dodatkowo, teoretycznej supresji produkcji zmutowanego białka przeciwdziała poimplantacyjne, postępujące odkładanie natywnego TTR,4,6 którego mechanizm nie jest do końca poznany. W rzeczywistości sercowe odkładanie TTR po TxH wpływa na zachorowalność i śmiertelność.
Potrzeba lepszego zrozumienia patogenezy ATTR i ograniczeń TxH pobudziła rozwój kilku leków.
Te nowe związki działają w różnych punktach kaskady amyloidogenezy TTR (ryc. 6). Leczenie zawsze będzie polegało na zmniejszeniu ilości białka prekursorowego, chociaż równie ważne będzie unikanie odkładania i eliminowanie istniejących odkładów. Dlatego uważamy, że w przyszłości podejście do tej choroby będzie miało formę leczenia skojarzonego.
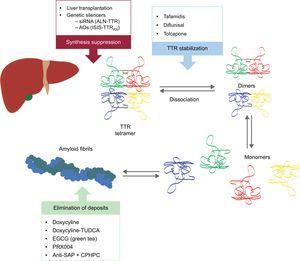
Szczególne terapie w amyloidozie transtyretynowej serca i główne cele. AntiSAP + CPHPC, antiserum amyloid P component + (R)-1–6-oxo-hexanoyl]pyrrolidine-2-carboxylic acid; AOs, antisense oligonucleotides; EGCG, epigallocatechin-3 gallate; siRNA, small interfering RNA; TTR, transtyretyna; TUDCA, tauroursodeoxycholic acid.
Supresja syntezy transtyretyny
Prowadzone są dwie linie badawcze nad hamowaniem ekspresji wątrobowej TTR: zastosowanie małego interferującego RNA (siRNA) oraz zastosowanie leków z grupy oligonukleotydów antysensownych (AO).
- –
SiRNA to dwuniciowe cząsteczki RNA, które wyciszają sekwencje messenger RNA poprzez specyficzne wiązanie się z nimi, co zapobiega tworzeniu się białek. Stwierdzono, że patisiran (ALN-TTR02) zmniejsza wytwarzanie TTR o 80%.38 U pacjentów z ATTRm redukcja TTR wyniosła 87%.39 W badaniu 2. fazy uzyskano obiecujące wyniki, wykazując stabilne parametry echokardiograficzne, czynnościowe i analityczne po 12 miesiącach i 24 miesiącach.40 Wyniki badania neurologicznego 3. fazy u pacjentów z ATTRm oraz subanalizy pacjentów z zajęciem serca spodziewane są w 2017 roku (tab. 2). Inny lek, revusiran (ALN-TTR01), jest podawany podskórnie i różni się od patisiranu nanocząsteczkami lipidowymi, które enkapsulują siRNA. Lek ten był przedmiotem badania klinicznego III fazy u pacjentów z ATTRm, którzy mają choroby serca. Badanie przerwano w ubiegłym roku z powodu nieoczekiwanego wzrostu śmiertelności w grupie leczonej (tabela 2).
Tabela 2.Główne trwające badania kliniczne w amyloidozie transtyretynowej serca
Mechanizm działania Związek Badanie Projekt. Pacjenci (N) i podtyp ATTR Interwencja Pierwszorzędowe punkty końcowe Sytuacja/wyniki Tłumienie syntezy TTR Patisiran (ALN-.TTR02) NCT01961921 Badanie II fazy, wieloośrodkowe 27
ATTRm (11 zajęcie serca)Patisiran 0.30 mg/kg IV co 3 tyg. przez 2 lata Długoterminowe bezpieczeństwo. Drugorzędowe punkty końcowe: wpływ na zaburzenia neurologiczne i parametry kardiologiczne Dobrze tolerowany lek, o podobnym profilu bezpieczeństwa w fenotypie neurologicznym i kardiologicznym
Troponina I, NT-proBNP i dane echokardiograficzne pozostały stabilne po 12 mo i 24 moNCT01960348 (APOLLO) Faza III, randomizowana, podwójnie ślepa, kontrolowana placebo, wieloośrodkowa 225
ATTRm z zajęciem układu neurologicznegoPatisiran infuzja IV vs placebo 2:1 Zmiany w mNIS+7 Oczekiwane listopad 2017
Subanaliza pacjentów z przewidywanym zajęciem sercaNCT02510261 APOLLO extension study Patisiran infusion IV vs placebo 2:1 przez 52 wk Bezpieczeństwo i długoterminowe działania niepożądane Trwające Revusiran (ALN-TTR01) NCT02319005 (ENDEAVOUR) Faza III, randomizowane, podwójnie zaślepione, kontrolowane placebo 206
ATTRm z zajęciem sercaRevusiran 500mg 5 d, następnie co tydzień przez 2 lata vs placebo Zmiany w teście 6-m marszu i w wartościach TTR w osoczu Zaniechanie z powodu zwiększonej śmiertelności w ramieniu z revusiranem ISIS-TTRRX NCT01737398 Faza II/III, randomizowany, podwójnie ślepy, kontrolowany placebo, wieloośrodkowy 172
ATTRm z neuropatią; 50% współistniejącego zajęcia sercaISIS-TTRRX 300mg SC co 12 h przez 1 wk, następnie co tydzień przez 64 wk vs placebo Zmiany w kwestionariuszu mNIS+7 i kwestionariuszu jakości życia Norfolk Przewidywany wrzesień 2017
Zgłoszono przypadki ciężkiej małopłytkowości i krwawienia
Analiza parametrów echokardiograficznych i NT-.proBNP oczekiwanych u pacjentów bez nadciśnienia tętniczego z LVH > 12 mmTerapia kardiomiopatii TTR za pomocą specyficznego dla TTR oligonukleotydu antysensownego Faza II, open-label, nonrandomized 20
ATTRm z zajęciem serca i ATTRwtISIS-TTRRX 300mg SC co 12 h/wk Parametry echokardiograficzne i MRI serca vs historyczne kontrole Brak pogorszenia odkształcenia i zmniejszenie masy LV o około 5%
6 pacjentów ukończyło 12 mo; 15 pacjentów 6 mo; 1 pacjent TxCNCT02627820 Phase II, open-label, nonrandomized 50
ATTRwtISIS-TTRRX 300mg SC co 12 h przez 1 wk, następnie 1 wk przez 18 wk Zmiany odkształcenia mierzone metodą śledzenia plamki Unieważnione bez rozpoczęcia rekrutacji pacjentów Badanie III fazy z zastosowaniem produktu ISIS-TTRRX w leczeniu kardiopatii amyloidowej TTR Badanie III fazy, randomizowane, podwójnie zaślepione, kontrolowane placebo, wieloośrodkowe 490
ATTRwt i ATTRm z zajęciem sercaISIS-TTRRX 300mg SC co 12 h przez 1 tyg, następnie co tydzień przez 16 tyg z placebo, następnie co tydzień przez 24 mo Śmierć, TxC, lub przyjęcie z przyczyn sercowo-naczyniowych On hold Stabilizacja TTR Tafamidis NCT01994889 Faza III, randomizowany, podwójnie ślepy, kontrolowany placebo, wieloośrodkowe 441
ATTRwt i ATTRm z zajęciem sercaTafamidis 20mg lub 80mg doustnie co 24 h przez 30 mo vs placebo Śmiertelność z wszystkich przyczyn i śmiertelność z przyczyn sercowo-naczyniowych oraz śmiertelność z przyczyn sercowo-naczyniowych.cause mortality and cardiovascular hospitalization Ends February 2018 NCT02791230 Extension Phase III NCT01994889 330
ATTRwt and ATTRm with heart involvementTafamidis 20 mg lub 80 mg doustnie co 24 h przez 60 mo All–.śmiertelność z wszystkich przyczyn i częstość występowania działań niepożądanych Przewidywany grudzień 2021 NCT00935012 Faza II, otwarte badanie skuteczności i bezpieczeństwa 31
ATTRwt lub ATTRm p.Val122Ile z zajęciem sercaTafamidis 20mg doustnie Bezpieczeństwo i skuteczność Trwające do grudnia 2021 Diflunisal NCT00294671 Faza III, randomizowane, podwójnie zaślepione, placebo-controlled, multicenter 130
ATTRm z fenotypem neurologicznym (50% z zajęciem serca)Diflunisal 250mg doustnie co 12 h vs placebo over 24 mo NIS+7 at 24 mo NIS+7 diflunisal vs placebo 16.3 (P Brak zmniejszenia grubości komory lub odkształcenia u pacjentów z zajęciem serca vs placebo Eliminacja złogów Doxycyklina + TUDCA/UDCA NCT01171859 Faza II, otwarta, nierandomizowana, prospektywna 40
ATTR (25 ATTRm, 13 ATTRwt i 2 biorców przeszczepu wątroby domino)Doxycyklina 100 mg co 12 h + TUDCA 250 mg co 8 h przez 12 mo, następnie 6 mo bez terapii Wzrost 14 pacjentów wycofało się
Niekorzystne reakcje skórne, 16 pacjentów
68% z 25 ocenianych pacjentów spełniło pierwszorzędowy punkt końcowy
Ogólna poprawa szczepu przy 12 mo i pogorszenie po 6 mo bez terapiiNCT01855360 Faza II, otwarta, nierandomizowana, prospektywna vs kontrole historyczne 30
Amyloidoza ATTR serca (27 ATTRwt i 3 ATTRm). Kontrole historyczne, 14 pacjentów z ATTRwtDoksycyklina 100 mg co 12 h + TUDCA 250 mg co 8h przez 18 mo Zmiany odkształcenia podłużnego co 6 mo 22 pacjentów ukończyło badanie i nadawało się do oceny
Większe pogorszenie odkształcenia w grupie kontrolnej vs grupie leczonej
Zwiększone NT-proBNP w grupie leczonej; nie mierzono w grupie kontrolnejNCT01677286 Faza II, badanie otwarte, nierandomizowane, prospektywna 25
Amyloidoza układowa (6 ATTRwt i 3 ATTRm)Doksycyklina 100 mg co 12 h przez 12 mo Bezpieczeństwo leku
Reakcja narządów dotkniętych chorobąPogorszenie NT-.proBNP i czynności nerek
Brak poprawy innych badanych parametrów
60% pacjentów miało powikłania skórne, a 30% wycofało się z powodu problemów skórnych lub żołądkowo-jelitowychNCT01171859 Faza II, otwarta, nierandomizowana, prospektywna 45
35 z zajęciem serca; 25 ATTRm; 5 ATTRm z TxH; 13 ATTRwt; i 2 biorców przeszczepu wątroby dominoDoksycyklina 100 mg co 12 h + TUDCA 250 mg co 8 h przez 12 mo
Kolejna faza obserwacji bez leczenia przez 6 moOdpowiedź na leczenie zdefiniowana jako Odpowiedź na leczenie kardiologiczne oceniona u 25 pacjentów
68% miało odpowiedź na leczenie kardiologiczne
Wzrost NT-.proBNP i pogorszenie odkształcenia podczas obserwacji bez leczenia
Wysoka liczba wycofań z powodu działań niepożądanych
14 wycofało się w fazie leczenia, a 5 przerwało w fazie bez leczeniaEffect of doxycycline + UDCA on ATTR Phase II, otwarta, nierandomizowana, prospektywne 28
ATTR z zajęciem serca (27 ATTRm i 1 ATTRwt)Doksycyklina 200 mg/d przez 4 tyg, następnie zawieszona 2 tyg, Następnie UDCA 750 mg/d przez 12 mo
Późniejsza faza kontrolna bez leczenia przez 6 moZmiany w NT-proBNP i punktacji Kumamoto Tylko 14% ukończyło badanie, a 36% ukończyło 12 mo
Brak zmian w NT-proBNP w 6 mo i pogorszenia w 12 mo
Stabilne LVH
Pogorszenie wyniku w skali Kumamoto w 12 moEGCG NCT01171859 Faza II, open-label, nonrandomized, prospective 25
ATTRwt600 mg, EGCG for 12 mo Changes in ECHO and cardiac MRI (n = 14) Decreased LV mass 6% by cardiac MRI (P = 0.03)
LVEF, grubość mięśnia sercowego i MAPSE wg ECHO bez zmianAntiSAP + CPHPC NCT03044353 Faza II, otwarta, randomizowana 40
Kohorta 1: sercowa amyloidoza ATTR
Kohorta 2: pierwotna amyloidoza po 6 mo chemioterapiiAnti-SAP + CPHPC co miesiąc przez 6 mo Zmniejszone obciążenie amyloidem według MRI serca i ECHO Start w 2017 roku AntiSAP + CPHPC, składnik antysurowicy amyloidu P + kwas (R)-1–6-okso-heksanoilo]pirolidyno-2-karboksylowy; ATTRm, zmutowana amyloidoza transtyretyny; ATTRwt, amyloidoza transtyretyny typu dzikiego; BNP, mózgowy peptyd natriuretyczny; ECHO, echokardiogram; EGCG, galusan epigallokatechiny-3; IV, dożylnie; LV, lewa komora; LVEF, frakcja wyrzutowa lewej komory; LVH, przerost lewej komory; MAPSE, skurczowe wychylenie płatka pierścienia mitralnego; mNIS, zmodyfikowany wynik upośledzenia neuropatii; MRI, rezonans magnetyczny; NIS, wynik upośledzenia neuropatii; NIS-LL, Neuropathy Impairment Score of the Lower Limbs; NT-proBNP, amino-terminal pro-brain natriuretic peptide; SC, podskórnie; TTR, transtyretyna; TUDCA, kwas tauroursodeoksycholowy; TxC, przeszczep serca; TxH, przeszczep wątroby; UDCA, kwas ursodeoksycholowy.
- –
AOs to krótkie pasma oligonukleotydów, które specyficznie wiążą się z RNA, uniemożliwiając translację i syntezę białek docelowych.4 ISIS-TTRRX jest podskórnym AO, z wykazanym zależnym od dawki zmniejszeniem wartości TTR o 75% do 90% u zdrowych ochotników.4 Badanie III fazy u pacjentów z ATTRm i fenotypem neurologicznym zakończyło się w marcu 2017 r., a jego wyniki spodziewane są do końca 2017 r. Jednak amerykańska Agencja Żywności i Leków odroczyła rozpoczęcie badania III fazy u pacjentów z ATTRwt i ATTRm z chorobą serca z powodu przypadków ciężkiej małopłytkowości w badaniu neurologicznym (Tabela 2). Ponieważ u 50% uczestników badania neurologicznego występowała choroba serca, wyniki tego substytucyjnego badania kardiologicznego zadecydują o ewentualnym wznowieniu badania III fazy. Z drugiej strony, istnieją wstępne dane z otwartego badania fazy II. W tym badaniu 22 pacjentów z ATTRwt i ATTRm z chorobą serca otrzymywało cotygodniową iniekcję ISIS-TTRRX. Według raportu profil bezpieczeństwa leku jest bardzo korzystny, a dane pośrednie dotyczące progresji choroby serca za pomocą CMR, NT-proBNP i testu 6-minutowego są pozytywne.41
Stabilizacja transtyretyny
Dysocjacja tetrameru TTR na podjednostki jest kluczowym etapem tworzenia fibryli ATTR. Diflunisal i tafamidis to 2 stabilizatory TTR o wykazanej skuteczności w polineuropatii ATTRm.
- –
Tafamidis jest podawaną doustnie małą cząsteczką, która wiąże się z TTR w miejscach wiązania T4, stabilizując białko i zapobiegając jego dysocjacji. Po opublikowaniu wyników randomizowanego badania przeprowadzonego metodą podwójnie ślepej próby u 125 pacjentów z ATTRm i mutacją Val30Met w początkowych stadiach choroby neurologicznej,42 Europejska Agencja Leków zatwierdziła jego stosowanie w 2011 roku jako sierocego produktu leczniczego w celu opóźnienia progresji neurologicznej. Najnowsze dane wskazują na skuteczność leku w osiąganiu stabilności neurologicznej u co najmniej 60% uczestników po ponad 4 latach obserwacji. Dotychczas lek ten ma ograniczone zastosowanie w ATTR i chorobach kardiologicznych. W badaniu II fazy u 21 pacjentów z ATTRm i różnymi mutacjami wykazano, że NT-proBNP i parametry echokardiograficzne pozostawały stabilne po 12 miesiącach.43 Dane z 5-letniego badania kohortowego potwierdziły, że lek był dobrze tolerowany w dawce 20 mg, chociaż niewielu pacjentów z ATTRwt pozostawało stabilnych po 3,5 roku.44 Badanie ATTR-ACT jest 30-miesięcznym badaniem III fazy oceniającym skuteczność, bezpieczeństwo i tolerancję dawek 20 mg i 80 mg tafamidisu w porównaniu z placebo u 440 pacjentów z ATTRm, ATTRwt i HF. Pierwszorzędowy punkt końcowy obejmuje śmiertelność i przyjęcie do szpitala. Jego wyniki spodziewane są w 2018 r.3,27
- –
Diflunisal jest niesteroidowym środkiem przeciwzapalnym, który stabilizuje cząsteczki TTR w warunkach in vitro. Nie jest on dostępny w Hiszpanii, ale może być wymagany medycznie z zagranicy w ramach compassionate use. W badaniu III fazy ATTRm u pacjentów z przewagą objawów neurologicznych, z których ponad połowa miała chorobę serca, nie stwierdzono istotnych różnic w parametrach echokardiograficznych w okresie badania (tab. 2).45 Potencjalne działania niepożądane ze strony przewodu pokarmowego, niewydolność nerek, zatrzymanie wody w organizmie i nadciśnienie tętnicze sprawiają, że nie jest to lek odpowiedni dla pacjentów z chorobą serca. Dowody dotyczące stosowania diflunisalu u pacjentów z ATTR są bardzo ograniczone. Istnieje jedno badanie, ale było ono ograniczone ze względu na nierandomizowany projekt jednoośrodkowy z niewielką liczbą pacjentów (n = 13) i krótką obserwacją. Nie było przyjęć z powodu zdekompensowanej HF, ale doszło do znacznego pogorszenia czynności nerek.46
- –
Ostatnio grupa hiszpańska wykazała, że tolkapon (doustny inhibitor katechol-O-metylotransferazy stosowany w leczeniu choroby Parkinsona) ma zdolność wiązania się in vitro z TTR pacjentów z ATTRwt i Val122Ile z większym powinowactwem niż inne stabilizatory.47
Eliminacja złogów amyloidu
Złogi amyloidu są bardzo stabilne i wydaje się, że organizm ludzki ma niewielką zdolność do ich eliminacji. Jednak leczenie, które zapobiega wytwarzaniu nowego amyloidu, takie jak chemioterapia w AL, może stopniowo eliminować złogi w różnym, specyficznym dla danego narządu tempie. Klirens sercowy jest szczególnie niski i jak dotąd dowody na regresję są skąpe. Obecnie trwają badania nad kilkoma cząsteczkami mającymi przyspieszyć usuwanie amyloidu z serca w ATTR:
- –
Doksycyklina (powszechnie stosowany antybiotyk) zaburza tworzenie się fibryli amyloidowych. Wykazano, że synergistyczne działanie skojarzone doksycykliny i kwasu żółciowego tauroursodeoksycholowego (TUDCA), stosowanego w leczeniu chorób wątroby, eliminuje złogi TTR w modelach zwierzęcych. W badaniu II fazy z udziałem 20 pacjentów nie wykazano progresji kardiologicznej ani neurologicznej po 1 roku leczenia doksycykliną/TUDCA, przy akceptowalnym profilu bezpieczeństwa i tolerancji.4 W innych badaniach II fazy próbowano potwierdzić te wyniki, stosując skojarzoną doksycyklinę/TUDCA, doksycyklinę/ kwasursodezoksycholowy lub samą doksycyklinę.48-50 Wstępne wyniki jednego z tych badań sugerują efekt ochronny, z mniejszym pogorszeniem czynności serca spowodowanym napięciem w grupie leczonej. W innym z tych badań uzyskano podobne wyniki u 40 pacjentów z ATTR: między innymi NT-proBNP, klasa czynnościowa, LVEF i parametry grubości mięśnia sercowego pozostały stabilne po 12 miesiącach (tab. 2). Niemniej jednak wszystkie te badania charakteryzowały się wysokim wskaźnikiem rezygnacji (35%-44%), głównie z powodu działań niepożądanych, w szczególności nadwrażliwości na słońce i dolegliwości żołądkowo-jelitowych (do 30%).48-50
- –
GEGCG (galusan epigallokatechiny-3) jest najobficiej występującą katechiną w zielonej herbacie i wykazano, że in vitro oraz w modelu mysim hamuje tworzenie amyloidu i eliminuje istniejące złogi.4 W badaniu CMRI wykazano, że codzienne podawanie 600 mg EGCG wiązało się ze stabilizacją masy lewej komory w grupie 25 pacjentów (Tabela 2).51
- –
PRX004 jest przeciwciałem monoklonalnym, które działa poprzez wiązanie się z epitopami specyficznymi dla monomeru i błędnie sfałdowanego TTR. W ten sposób wywołuje eliminację złogów poprzez aktywację fagocytozy.52 Podstawa jego mechanizmu działania jest podobna do mechanizmu działania przeciwciała stosowanego w AL. Badania II fazy nad tym przeciwciałem przynoszą obiecujące wyniki. Badanie fazy I-II tego nowego przeciwciała ma się rozpocząć w 2017 r.
- –
Niezależnie od rodzaju białka prekursorowego amyloidu, wszystkie złogi zawierają składnik P amyloidu surowicy (SAP). Wykorzystując tę cząsteczkę jako cel, wykazano, że przeciwciała anty-SAP wywołują reakcję zależną od makrofagów i dopełniacza, która spowodowała znaczną eliminację trzewnych złogów amyloidu w modelu mysim. Związek bis-D-proliny CPHPC może neutralizować SAP w osoczu, a jednoczesne podawanie z anty-SAP IgG umożliwia przeciwciałom dotarcie do złogów zawierających SAP w tkankach.53 W badaniu I fazy opublikowanym w 2015 r. wykazano eliminację złogów wątrobowych u 15 pacjentów z amyloidozą układową bez zajęcia serca, przy niewielkiej liczbie działań niepożądanych.53 W 2017 roku ma się rozpocząć badanie II fazy z udziałem pacjentów z amyloidozą sercową ATTR i AL (tab. 2).
WNIOSKI
Transtyretynową amyloidozę sercową rozpoznaje się coraz częściej. Scyntygrafia 99mTc-DPD i CMRI są przykładami technik, które mogą być wykorzystane do prostej i wczesnej identyfikacji pacjentów z ATTR.
Kilka leków specyficznych dla ATTR znajduje się obecnie w końcowych fazach badań. Dlatego wierzymy, że amyloidoza sercowa ATTR zostanie wkrótce uznana za jednostkę uleczalną, a nie chorobę śmiertelną.
FINANSOWANIE
Praca ta została przeprowadzona przy częściowej pomocy Instytutu Zdrowia Carlosa III oraz Hiszpańskiego Towarzystwa Kardiologicznego (grant badawczy 2016 dla E. González-López). Pomoc Instytutu Zdrowia Carlos III jest finansowana ze środków Europejskiego Funduszu Rozwoju Regionalnego „Another Way to Make Europe”.
KONFLIKTY INTERESÓW
E. González-López brał udział jako prelegent w działaniach organizowanych przez firmę Pfizer. P. Garcia-Pavia otrzymał płatności jako prelegent w działaniach organizowanych przez firmę Pfizer oraz jako konsultant firm Alnylam, Prothena i Pfizer. E. González-López, A. López-Sainz i P. Garcia-Pavia deklarują, że firma Pfizer finansowała projekty badawcze prowadzone przez ich instytucje.
.