To są notatki z wykładu 4 kursu biologii komórki Harvard Extension.
Szlak wydzielniczy odnosi się do retikulum endoplazmatycznego, aparatu Golgiego i pęcherzyków, które przemieszczają się między nimi, jak również do błony komórkowej i lizosomów. Nazwano ją „wydzielniczą”, ponieważ jest to droga, za pomocą której komórka wydziela białka do środowiska pozakomórkowego. Ale jak zwykle, etymologia mówi tylko ułamek historii. Szlak ten przetwarza również białka, które będą związane z błoną (czy to w błonie komórkowej, czy w samych błonach ER lub Golgiego), jak również enzymy lizosomalne, a także wszelkie białka, które będą żyły swoim życiem w samym szlaku wydzielniczym. Robi też kilka rzeczy innych niż przetwarzanie protein.
Cytozol i „lumen” (ciecz, która wypełnia drogę wydzielniczą) to różne środowiska chemiczne, i zwykle nigdy się nie mieszają. Cytozol jest redukcyjny (kiedy jesteś w cytozolu, ciągle spotykasz cząsteczki, które chcą zaoferować ci elektrony), a ER, Golgi i środowisko pozakomórkowe są oksydacyjne (cząsteczki ciągle podchodzą do ciebie prosząc o elektrony). Zobacz redoks, jeśli nadal jesteś zdezorientowany. To sprawia, że warunki składania białek są różne: na przykład, wiązania disulfidowe zwykle tworzą się tylko w warunkach oksydacyjnych. Co więcej, różne białka mogą żyć tylko w szlaku wydzielniczym lub tylko w cytozolu. Ścieżka wydzielnicza zapewnia komórce drogę dla rzeczy, które nie powinny znajdować się w cytoplazmie i/lub są najbardziej użyteczne, gdy są skoncentrowane w wyspecjalizowanym przedziale z ich pożądanymi partnerami. Hepatocyty (w wątrobie) sekwestrują leki i toksyny w gładkim ER i tam je rozkładają w celu wydalenia z organizmu. Ścieżka wydzielnicza nie jest przylegająca, ale każdy ruch między jej składnikami jest w małych pęcherzykach-off mikrokosmosów własnego świata chemicznego, zwanych pęcherzykami.
Wiele białek, które przechodzą przez ścieżkę wydzielniczą, nigdy nie dotyka cytozolu – z wyjątkiem części białek błonowych, które wystają po stronie cytozolowej. Wiele z nich potrzebuje chaperonów, które pomogą w ich składaniu, i/lub całej serii modyfikacji potranslacyjnych, aby były gotowe do pełnienia swojej natywnej funkcji, a szlak wydzielniczy specjalizuje się w dostarczaniu im tego wszystkiego.
Dzisiejszy wykład skupi się na tym, jak białka są translokowane do ER i jak podróżują (w pęcherzykach) pomiędzy ER, Golgiego i innymi miejscami przeznaczenia. Jest to pięknie przedstawione w filmie Life of the Cell:
Siateczka endoplazmatyczna jest pierwszym etapem w szlaku wydzielniczym. Jego błona jest ciągła z zewnętrzną błoną jądrową, choć nie jest jasne, dlaczego to ma znaczenie, ponieważ nie jest tak, że białka zaczynają swoje życie w jądrze. Raczej mRNA dryfuje w cytoplazmie, dopóki nie zostanie wychwycone przez rybosom zainteresowany jego translacją. W „translokacji potranslacyjnej” nowe białko po przetłumaczeniu jest przenoszone do ER. W bardziej interesującym zjawisku zwanym „translokacją kotranslacyjną” rybosom rozpoczyna translację tak jak każde inne białko, ale gdzieś w pierwszych 16 do 30 aminokwasach trafia na peptyd sygnałowy (aka sekwencja sygnałowa). Motywem tego sygnału jest często 1 dodatnio naładowany aminokwas, po którym następuje 6-12 aminokwasów hydrofobowych. Motyw ten zostaje rozpoznany przez cząsteczkę rozpoznającą sygnał (SRP – rybonukleoproteina, czyli hybrydowa cząsteczka RNA/białko), która wiąże się z nim i uniemożliwia rybosomowi kontynuowanie translacji. Translacja zostaje zatrzymana do momentu, gdy kompleks rybosom/SRP napotka receptor SRP na błonie ER. Kiedy się spotkają, SRP i jego receptor wiążą po jednej cząsteczce GTP w błonie ER, co najwyraźniej wzmacnia ich wzajemne oddziaływanie. Szczęśliwie, wszystko to dzieje się w pobliżu translokonu Sec61 – kompleksu białkowego, który tworzy kanał przecinający błonę ER. Translokon jest tak naprawdę kompleksem trzech różnych białek (geny: SEC61A1 lub SEC61A2, SEC61B, SEC61G), z których podjednostka Sec61a posiada 10 a-helikali rozprzestrzeniających się po błonie i tworzących kanał. Po zadokowaniu rybosomu w błonie kontynuuje on translację, przepychając peptyd sygnałowy, a w końcu całe białko przez kanał do światła ER. Kiedy translacja się zatrzymuje, SRP i receptor SRP hydrolizują swoje GTP, aby uwolnić siebie nawzajem i ładunek rybosomu (musi to wymagać energii GTP, ponieważ pierwotne wiązanie było w dół), peptydaza sygnałowa rozszczepia peptyd sygnałowy z powstającego białka, a białko jest wolne, aby rozpocząć składanie w ER.
Kilku innych graczy jest zaangażowanych w niektóre białka ER. Transferaza oligosacharydowa, która dodaje grupy glikozylowe do asparagin w powstającym białku, jest częścią kompleksu translokalnego i faktycznie wykonuje glikozylację, podczas gdy nowe białko jest nadal tłumaczone. Tak więc, chociaż nazywamy glikozylację „modyfikacją potranslacyjną”, w tym przypadku jest ona faktycznie przeprowadzana podczas translacji. Ponadto, aby uzyskać właściwą strukturę, niektóre białka muszą być w pełni przetłumaczone, zanim będzie można rozpocząć ich składanie – jeśli pozwolono by N-końcowej części białka rozpocząć składanie od razu po wejściu do światła, miałoby ono niewłaściwą strukturę ogólną. Aby temu zapobiec, czasami BiP – chaperon wiąże białko, aby utrzymać je przez pewien czas w stanie rozłożonym. Wyobraźmy sobie BiP jako kolejnego Pac-Mana, który przyciska białko, aby utrzymać je w formie liniowej, jak Hsc70 w procesie ukierunkowania mitochondrialnego (patrz ostatni tydzień).
Tutaj filmik na ten temat:
Pierwsze kilka minut pokazuje podstawowy scenariusz opisany powyżej. Następnie przechodzi do bardziej złożonego scenariusza, który przedstawię za chwilę. FYI, wideo przedstawia dwie „kontrowersyjne” rzeczy nie zawarte w powyższym opisie: (1) peptyd sygnałowy ulegający degradacji w błonie, oraz (2) „białko wtykowe”, które zatrzymuje kanał przed/po translacji. Nie wszyscy naukowcy zgadzają się na te dwie rzeczy yet.
Wszystkie białka, które znamy przechodzą przez ścieżkę wydzielniczą zostały tam wskazane przez ludzi robiących eksperymenty lokalizacyjne, aby zobaczyć, gdzie w komórce leży białko. Dziwnym faktem dotyczącym ER jest to, że można umieścić komórkę w mikserze, a następnie ER po prostu zacznie łączyć się ze sobą, tworząc małe „mikrosomy”, które nie są przymocowane do jądra, ale tworzą przylegające pęcherzyki ER. Wtedy możesz zacząć grać w gry z proteazami – które rozkładają białka – i detergentami – które rozpuszczają błonę ER. Zakładając, że interesujące nas białko jest translokowane, można sprawdzić, czy (1) przetrwa traktowanie proteazą, ale (2) nie przetrwa traktowania proteazą + detergentem, czy jest to białko szlaku sekrecyjnego. Logika jest taka, że w przypadku (1) było ono chronione wewnątrz ER, ale w przypadku (2) rozpuściłeś ER, więc zostało zjedzone przez proteazę. Wszystko to zakłada, że masz przeciwciało lub jakiś inny sposób wykrywania, czy białko zainteresowania jest tam po tych zabiegach.
Ludzie również używali takich technik, aby dowiedzieć się, że tylko 70 aminokwasów nowego białka może być przetłumaczone, zanim będzie za późno, aby to białko skończyło w ER. Pamiętajmy, że peptyd sygnałowy znajduje się w pierwszych 16-30 aminokwasach, a translokacja do ER zależy od obecności SRP. Rybosomy tłumaczą w przewidywalnym tempie, więc ludzie dostali rybosomy zaczęły się na tłumaczenie niektórych mRNA, a następnie czekał ustalone ilości czasu przed dodaniem SRP, aby zobaczyć, ile tłumaczenie może wystąpić, zanim SRP nie może już zrobić jego pracy.
Receptor SRP i Sec61 białka są ER białek membranowych – i tam wiele innych ER błony, błony Golgiego i lizosomów białek membranowych, jak również. W rzeczywistości, nawet białka błonowe (patrz klasa 02) błony komórkowej są przetwarzane w szlaku wydzielniczym. Wiele z nich ma kilka lub kilkadziesiąt domen transmembranowych (20-25 hydrofobowych aminokwasów każda), które muszą być wstawione w odpowiedniej kolejności i orientacji (na przykład, naprawdę chcesz, aby twoje kanały jonowe i transportery skierowane we właściwym kierunku, do vs. poza komórkę). W związku z tym istnieje cała masa wymyślnych mechanizmów biologicznych, dzięki którym białka te są prawidłowo wprowadzane do błony. To jest to, co druga połowa powyższego filmu przedstawia.
Więc tutaj jest tautologia: niektóre białka mają sekwencję topogeniczną, która określa ich orientację w błonie. Sekwencja ta składa się z dwóch rodzajów sekwencji sygnałowych:
- Sekwencja stop-transfer (z jakiegoś powodu skracana STA) to 22-25 hydrofobowa sekwencja aminokwasowa gdzieś w środku białka, która tworzy helisę alfa. Po napotkaniu zostaje ona wepchnięta do błony, a następnie tłumaczenie reszty białka jest kontynuowane w cytosolu. Więc to rodzaj „cofnięcia” translokacji do ER, która została rozpoczęta przez peptyd sygnałowy na początku (N terminus) białka.
- sekwencja kotwicy sygnałowej (w skrócie SA) jest również 22-25aa hydrofobową helisą alfa, ale z serią ~3 dodatnio naładowanych aminokwasów po jej lewej lub prawej stronie. Podobnie jak peptyd sygnałowy, jest ona rozpoznawana przez SRP, który doprowadza rybosom do ER. Jednak w przeciwieństwie do peptydu sygnałowego, ta sekwencja alfa helikalna zostanie wprowadzona do błony ER. Orientacja wstawienia jest określona przez 3 dodatnio naładowane aminokwasy. Dodatnie ładunki muszą zawsze kończyć się po stronie cytozolowej, więc jeśli pojawiają się po (tj. C-końcu) sekwencji hydrofobowej, białko kończy się z jego C-końcem skierowanym do cytozolu, ale jeśli pojawiają się przed (tj. N-końcem) sekwencji hydrofobowej, białko kończy się z jego N-końcem skierowanym do cytozolu.
Z tych dwóch sygnałów jako bloki konstrukcyjne, można sobie wyobrazić białko z serii przystanku transferu i sygnał sekwencji kotwicy do tworzenia całej serii tam i z powrotem transmembranowych domen zszyte do błony, jak gdyby przez maszynę do szycia. Ludzie sklasyfikowali białka błonowe na pięć kategorii:
- Typ I ma tylko peptyd sygnałowy, a następnie jeden transfer stopu w środku. Dlatego kończy się z jego (hydrofilowy) N terminus w lumen, jego (hydrofobowy) środek w błonie i jego (hydrofilowy) C terminus w cytosol.
- Typ II nie zaczyna się od peptydu sygnałowego. Zaczyna się jak każde inne białko, ale w środku ma sekwencję kotwicy sygnałowej z aminokwasami +++ przychodzącymi jako pierwsze i serią hydrofobową po nich. To sprawia, że białko ulega translokacji w połowie drogi translacji, przy czym już translokowana część N-końcowa wystaje do cytozolu (ponieważ +++ muszą pozostać cytozolowe), a zaczynająca się translokować część C-końcowa jest translokowana bezpośrednio do ER. Tak więc kończy się to transmembraną z jej C-końcem w ER i N-końcem w cytosolu – odwrotnie niż w typie I.
- Typ III jest jak typ II – brak peptydu sygnałowego, tylko kotwica sygnałowa w środku, ale w tym przypadku +++ pojawiają się po sekwencji hydrofobowej, co odwraca orientację. Tak więc kończy się to tym, że jego N-końcówka znajduje się w ER, a C-końcówka w cytozolu. Przeciwieństwo typu II i, w końcu, to samo co typ I, choć dostał się tam w inny sposób – nie ma peptydu sygnałowego, który ulega rozszczepieniu w ER.
- Białka typu IV lub „wieloprzebiegowe” mają naprzemienną serię sekwencji sygnałowych i sekwencji transferu stopu. Są to wyraźnie więcej niż jeden „typ”, ale nie są prawie tak zróżnicowane, jak twoja kombinatoryczna wyobraźnia może pozwolić. Orientacja pierwszej sekwencji sygnałowej określa, czy terminus N znajdzie się w cytozolu czy w ER, a całkowita liczba sekwencji stop-transfer + kotwica sygnałowa określa, gdzie znajdzie się terminus C: parzysta liczba = po tej samej stronie co terminus N, nieparzysta liczba = po przeciwnej stronie niż terminus N. Sekwencje STA i SA muszą się ściśle przeplatać, z wyjątkiem tego, że można zacząć od dwóch sekwencji kotwicy sygnałowej, jeśli pierwsza z nich jest zorientowana z terminusem N do cytozolu. Aby wykpić ten schemat kategoryzacji, ludzie zdefiniowali kilka niekompletnie zdefiniowanych podtypów typu IV, gdzie typ IVa jest N-końcowy w cytozolu (więc zaczyna się jak białko typu II), a typ IVb jest N-końcowy w świetle (zaczyna się jak białko typu III, ale potem ma jeszcze jedną sekwencję SA, która umieszcza go z powrotem w ER). GLUT1 z klasy 02 jest typem IVa.
- Białka zakotwiczone w GPI, które są piątym typem, ale nie są nazywane typem V, zaczynają się od peptydu sygnałowego i kończą się hydrofobowym C-terminusem, który pozostaje osadzony w błonie. Ten hydrofobowy koniec zostaje odł±czony i zast±piony przez GPI, który również pozostaje osadzony w błonie. PrP jest jednym z nich – więcej na ten temat później.
Do tej pory omówiliśmy, jak białka mogą skończyć w świetle ER lub obejmując błonę ER. Większość białek opuścić ER w ciągu kilku minut, transportowane w pęcherzykach związanych do Golgiego, a następnie później do wydalania, lizosomów lub błony komórkowej. Że do przodu kierunek podróży nazywa anterograde; cofanie się od Golgiego do ER jest retrograde transport.
Oba rodzaje transportu odbywają się w pęcherzykach związanych z błoną. Te pączkują z błony skądkolwiek pochodzą, a później łączą się z błoną gdziekolwiek zmierzają – pięknie przedstawione na ~2:25 w filmie Life of the Cell powyżej. Ciało, z którego tworzą się pęcherzyki jest „przedziałem dawcy”, a miejsce docelowe, do którego później fuzja jest „przedziałem akceptora”.
Proces pączkowania wymaga, aby białka G w błonie rekrutowały białka płaszcza. W szczególności, dla transportu anterogradowego, białko G Sar1 (gen: SAR1A) rekrutuje COPII („cop two”); dla transportu retrogradującego, białko G ARF rekrutuje COPI (wymawiane jako „cop one”). Te białka G są aktywowane do wykonania tej pracy, gdy GEF ładuje je GTP, zamieniając GDP.
Więc kroki w transporcie anterogradowym, na przykład, są następujące:
- Sec12-GEF (Sec oznacza wydzielanie) ładuje Sar1 z GTP. Kiedy Sar1 jest związany z GDP, po prostu pływa po przedziale donorowym, ale kiedy jest związany z GTP, ulega zmianie konformacyjnej, która powoduje, że jego inaczej spalony N-końcowy hydrofobowy ogon wystaje, dzięki czemu wbija się w błonę, gdzie białka COPII zaczynają się gromadzić, ponieważ naprawdę lubią ten ogon.
- COPII zaczynają polimeryzować i ze względu na swoją konformację mają wewnętrzną preferencję do zakrzywienia, więc ich gromadzenie się zaczyna sprawiać, że pączkowanie się dzieje. W tym samym czasie białka zwi±zane z błon±, które musz± być transportowane – zidentyfikowane przez sekwencję aminokwasow± DXE (tj. asparaginian-anything-glutamate) tworz±c± miejsce wi±ż±ce w ich czę¶ci cytozolowej – zostaj± zrekrutowane do nowo tworz±cej się cz±steczki. Białka związane z błoną działają jak receptory, rekrutując białka lumenalne, które są związane z Golgim, do zawieszenia się we wklęsłej przestrzeni, gdzie trafią do pęcherzyka po jego uformowaniu.
- Gdy już wystarczająco dużo COPII przybyło, pęcherzyk odrywa się, w którym to momencie Sar1 hydrolizuje GTP, dostarczając energii, aby wessać swój hydrofobowy ogon z powrotem do siebie, odcinając COPIIs luźne. Cząsteczka jest teraz odłączona od przedziału donora.
- Teraz, ze słabo wyjaśnionych (lub słabo zrozumianych?) powodów, płaszcz COPIIs po prostu rozpada się, odsłaniając receptory pod płaszczem, które kierują ukierunkowaniem cząsteczki. Gdy cząsteczka dociera do miejsca przeznaczenia, Rab-GTP wbudowany w błonę cząsteczki wchodzi w interakcję z efektorem Rab wbudowanym w błonę przedziału akceptorowego. Następuje wymiana spojrzeń, wzbudzane jest zainteresowanie. Wkrótce cząsteczka połączy się z błoną.
- Białka SNARE obecne zarówno na cząsteczce, jak i na błonie docelowej (odpowiednio V-SNARE i T-SNARE) oddziałują ze sobą, aby jeszcze bardziej zbliżyć błony. W tym przykładzie rozważymy VAMP (geny VAMP_) jako V-SNARE oraz Syntaksynę (geny STX__) i SNAP25 (gen SNAP25) jako T-SNARE. Syntaksyna i SNAP25 są białkami błonowymi; Syntaksyna ma 1 helisę alfa, a SNAP25 ma 2, wszystkie po stronie cytozolowej. Helisy alfa napędzają interakcję z VAMP. Helisy alfa przeciwległych stron mają bardzo duże powinowactwo do siebie, co powoduje, że błony zbliżają się do siebie na tyle, że dochodzi do fuzji. Kiedy już do tego dojdzie, ponowne rozerwanie V-SNARE i T-SNARE wymaga dwóch białek: NSF (gen: NSF; skrót od NEM sensitive factor) i alfa-SNAP (gen: NAPA), rozpuszczalne białko przyłączeniowe NSF. NSF jest ATPazą i spala ATP, aby napędzać energetycznie pod górę demontaż kompleksu.
Teraz o transporcie wstecznym. Dlaczego w ogóle istnieje transport wsteczny? Oto niewyczerpująca lista niektórych powodów:
- Niektóre białka błonowe zaczynają swoje życie w ER, muszą zostać zmodyfikowane w Golgi, ale potem muszą wrócić do ER. Robią to za pomocą sekwencji aminokwasowej KKXX.
- Jest też sekwencja aminokwasowa KDEL na końcu C niektórych białek lumenalnych, która ma za zadanie utrzymać je w ER, ale nie jest idealna – czasami kończą w Golgim, w którym to przypadku są kierowane z powrotem do ER przez transport wsteczny zależny od sekwencji KDEL dla rozpoznania. Mechanizm jest całkiem zgrabny – białka, które rozpoznają i wiążą się z KDEL, robią to tylko przy niskim pH, a pH Golgiego jest niższe niż ER, więc wiążą KDEL w Golgim, a następnie uwalniają go, gdy są z powrotem w bardziej neutralnym pH ER.
- Pomyśl o tym, wszystkie białka, które uczestniczą w transporcie anterograde – V-SNARES, Rab, itp. – muszą wrócić do ER, aby mogły zrobić to wszystko od nowa, tak jak autobus musi wrócić do zajezdni autobusowej na koniec dnia.
- Jak zobaczymy wkrótce, Golgiego przychodzą w wielu etapach, które zależą od dodania enzymów z dalszego downstream.
Proces transportu wstecznego nie różni się tak bardzo od anterograde. Wykorzystuje ARF zamiast Sar1, COPI zamiast COPII, ale działa tak samo: ARF obciążony GTP pozwala swojemu hydrofobowemu ogonowi wbić się w błonę, przyciągając uwagę COPI. COPI ma dwa składniki, COPIalfa i COPIbeta, z których oba oddziałują z sekwencją KKXXX w celu rekrutacji białek związanych z błoną, przeznaczonych do transportu wstecznego. Niektóre białka mają również sekwencję RR (gdziekolwiek w białku), która może oznaczać je do transportu wstecznego.
Aparat Golgiego nie jest przylegający. Jest to ułożony w stos zestaw oddzielnych subkompartmentów zwanych sacs lub cisternae. Różne przedziały mają różne właściwości i białka odwiedzają je w określonej kolejności. W kolejności od ER do błony komórkowej, przedziały Golgiego są nazywane cis, medial, trans i trans-Golgi network. Każdy przedział ma różne enzymy, które modyfikują białka, a modyfikacje muszą się zdarzyć w pewnym porządku, stąd potrzeba ułożonego zestawu compartments.
Ale jak białka dojrzewają w Golgiego, to nie jest tak, że pączkują w pęcherzykach z jednego przedziału i przenieść do następnego. Raczej przedział, w którym już są, przemieszcza się na zewnątrz i „dojrzewa”, gdy nowe enzymy są do niego dodawane (z dalszej części łańcucha Golgiego) poprzez transport wsteczny. Dziwne, prawda? To trochę tak, jakbyś zamiast przechodzić ze szkoły podstawowej do gimnazjum do liceum po prostu pozostał w jednym budynku szkolnym przez całe swoje dzieciństwo i dorastanie, a oni po prostu przynosili nowe podręczniki i nauczycieli każdego roku, aby utrzymać go odpowiednim do stopnia, który ty i twoi koledzy z klasy teraz osiągnęli. Oto jak wyglądają Golgiego jak się poruszają i ewoluują:
Więc jest (mało lub) nie ma transportu anterograde w Golgiego, ale mnóstwo transportu retrograde, aby przynieść każdą nową rundę enzymów w. Kiedy białka w końcu zakończyły pełny program nauczania K-12 w sieci Golgiego, przechodzą transport, aby przejść do ich ostatecznego destinaton. Pączkują w pęcherzyku, który pójdzie w jedno z trzech miejsc:
- Egzocytoza – fuzja z błoną komórkową. W ten sposób białka lumenalne będą wydzielane zewnątrzkomórkowo, a białka błonowe staną się białkami błony komórkowej.
- Pęcherzyki wydzielnicze – te po prostu trzymają się jako pęcherzyki w komórce, aż będą potrzebne – gdzie „potrzebne” oznacza, że ostatecznie ulegają egzocytozie. W neuronach, jest to miejsce, gdzie neuroprzekaźniki są przechowywane do momentu, gdy potencjał czynnościowy wymaga ich wydzielenia do synapsy. W żołądku komórki produkujące enzymy żołądkowe przechowują te enzymy w pęcherzykach wydzielniczych, dopóki spożycie pokarmu nie spowoduje ich uwolnienia do żołądka.
- Lizosomy – gdzie źle złożone białka ulegają degradacji.
Transport z sieci trans-Golgiego do tych miejsc docelowych różni się od innych transportów omówionych powyżej i często wiąże się z udziałem klathryny (geny CLT__). Pączkujące cząsteczki mają dwuwarstwową powłokę, z kompleksami białek adaptorowych (AP) jako warstwą wewnętrzną i klatyną jako warstwą zewnętrzną. Białka adaptorowe posiadają sygnał docelowy z motywem YXXh (h = Φ = dowolny aminokwas hydrofobowy). Klatyna tworzy tzw. formację 'klatyna-triskelion’ pokazaną tutaj:

(Image thanks to Wikimedia Commons user Phoebus87)
Klatyna jest również odpowiedzialna za endocytozę – odrywanie się pęcherzyków z materiałami pozakomórkowymi (i białkami błony komórkowej), aby dostać się do komórki. Nazywa się to endocytozą wspomaganą przez klathrynę. Receptory w błonie komórkowej ulegają endocytozie bardzo często: cała populacja receptorów hormonalnych zmienia się mniej więcej co godzinę, zwłaszcza gdy hormony są przyjmowane. Pobranie receptora do pęcherzyka jest jednym ze sposobów odcięcia przez komórkę przychodzącego sygnału do czasu, gdy będzie on mógł zostać przetworzony.
W notatkach dotyczących błony plazmatycznej krótko omówiono mukowiscydozę: CFTR jest transporterem ABC odpowiedzialnym za wypompowywanie Cl- z komórki (wpuszcza również Na+). Mutanty z utratą funkcji nie pompują Cl-, co usuwa siłę napędową osmozy, zagęszczając śluz i powodując problemy z oddychaniem. Istnieje co najmniej 127 różnych mutacji utraty funkcji CFTR (przynajmniej na tyle Natera przeprowadza testy), które (jeśli oba allele są upośledzone) powodują mukowiscydozę. Najczęstszą mutacją jest ΔF508, która stanowi ~3% wszystkich europejskich alleli CFTR i około 70% zmutowanych. Utrata tej jednej fenyloalaniny zmienia konformację CFTR w taki sposób, że dwu-kwasowy kod wyjścia (aminokwasy D565 i D567), który kieruje CFTR do pęcherzyków egzocytotycznych, nie jest już prawidłowo eksponowany i białko nigdy nie trafia do błony komórkowej.
Sekcja dyskusyjna
W sekcji czytamy Hu 2009, który wykazał, że białka atlastyny są zaangażowane w tworzenie rurkowej sieci ER. Dowody pochodziły prawie w całości z interakcji białko-białko. Byłem zaskoczony, że ten papier to wielka sprawa, ponieważ było milion prac pokazujących interakcje białko-białko dla huntingtyny, a nikt tak naprawdę nie wierzy we wszystkie z nich i niekoniecznie przybliżyło nas to do poznania, co robi huntingtyna lub co idzie nie tak w chorobie Huntingtona. Ale najwyraźniej Hu był w stanie przedstawić całkiem czysty przypadek interakcji atlastin z retikulonami jako sugerujący rolę w tworzeniu ER. Pomaga to, że Hu był w stanie pokazać „interakcję genetyczną” oprócz fizycznej (wiążącej) interakcji. Interakcja genetyczna” (musiałem to sprawdzić) oznacza, że „Czasami mutacje w dwóch genach wytwarzają fenotyp, który jest zaskakujący w świetle indywidualnych efektów każdej mutacji. To zjawisko, które definiuje interakcję genetyczną, może ujawnić funkcjonalne związki między genami i szlakami.” .
PrP
To jest dekada, więc niektóre rzeczy mogą być przestarzałe, ale znalazłem Harris 2003 (ft) przegląd biologii komórki PrP niezwykle jasny i pomocny. Kim & Hegde 2002 był też pomocny. PrP jest białkiem szlaku wydzielniczego. Jego pierwsze 22 aminokwasy (MANLGCWMLVLFVATWSDLGLC) to peptyd sygnałowy, który powoduje kotranslacyjną translokację do ER. Normalnie PrP wiąże się z GPI na swoim C-końcu i jest zakotwiczony na egzoplazmatycznej stronie błony. Ale aminokwasy 111-134 (HMAGAAAGAVVGGLGYMLGSAM) są rodzajem słabej sekwencji kotwicy sygnałowej (typ II, z aminokwasami +++ występującymi przed kotwicą sygnałową), która czasami, ale nie zawsze, staje się domeną transmembranową, odwracając terminus C do światła. Jeszcze bardziej mylące jest to, że sekwencja ta może czasami kończyć się jako domena transmembranowa bez inwersji, tak że N-końcówka znajduje się w świetle. Tak więc istnieją trzy topologie błonowe PrP: regularna stara GPI-zakotwiczona, i dwie orientacje transmembranowe, jak przedstawiono w Harris 2003 Fig 3:
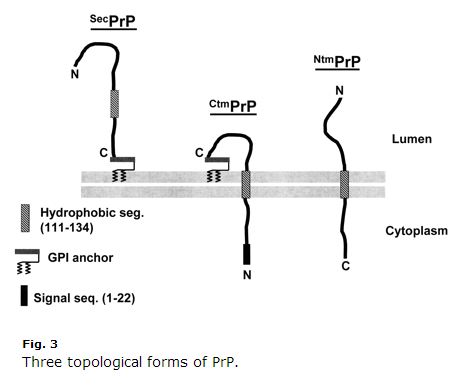
Zauważ jak dziwny jest CtmPrP. Jest transmembranowy, ale także zakotwiczony w GPI, a N-końcowy peptyd sygnałowy nigdy nie jest odłączany. Normalnie, formy transmembranowe stanowią < 10% całkowitej PrP. W niektórych warunkach laboratoryjnych odsetek ten jest wyższy, a dwie z mutacji powodujących GSS (A117V i P105L) również zwiększają frakcję CtmPrP do 20-30% wszystkich PrP. Z tych trzech form, istnieje duża ilość dowodów, że CtmPrP jest toksyczny, i że może odgrywać rolę w tworzeniu prionów, chociaż większość genetycznych mutacji chorób prionowych (w tym FFI D178N) nie wydaje się wpływać na topologię błony PrP lub frakcję CtmPrP.
Po tym jak PrP przechodzi przez Golgiego, jest kierowany do błony komórkowej. Ale według Harrisa, nie tylko tam siedzi – często przez endocytozę wspomaganą klatyną i cyklicznie przechodzi przez komórkę co ~60 minut, przy czym niektóre cząsteczki są rozszczepiane na każdym cyklu. Miedź stymuluje tę endocytozę PrP. Większość mutacji genetycznych chorób prionowych zmienia lokalizację PrP – zwykle, gdy mutacja jest obecna, mniej PrP znajduje się na powierzchni komórki, a więcej gromadzi się w ER.
.