INTRODUCTION
L’amylose est une maladie de dépôt causée par l’accumulation extracellulaire de fibrilles dont la source est constituée de protéines à structure instable qui se replient, s’agrègent et subissent un dépôt1. Ce dépôt peut altérer la structure des tissus et altérer la fonction de divers organes et systèmes.2
Les fibrilles amyloïdes sont insolubles et résistantes à la protéolyse et sont typiquement colorées par le rouge Congo, montrant une biréfringence jaune-vert intense sous lumière polarisée.3. Plus de 30 protéines peuvent provoquer un dépôt amyloïde, mais seulement 5 provoquent un dépôt significatif dans le tissu cardiaque1:
- –
Les chaînes légères, qui provoquent l’amylose primaire (AL).
- –
La transthyrétine (TTR), qui provoque l’amylose TTR (ATTR).
- –
Apolipoprotéine A.
- –
Fibrinogène.
- –
Protéine A amyloïde sérique, qui produit une amylose secondaire.
L’amylose primaire et l’ATTR sont les formes les plus courantes d’amylose cardiaque, la forme AL étant historiquement considérée comme plus fréquente dans les pays développés3.
La plupart des informations sur l’amyloïdose cardiaque ont été basées sur la forme AL. Cependant, bien que le nombre de patients atteints d’AL soit resté stable, le nombre de diagnostics d’ATTR a récemment augmenté et on pense maintenant que l’ATTR pourrait être beaucoup plus répandu que l’AL.2
L’amylose à transthyrétine a très souvent fait l’objet de diagnostics erronés ou de retards importants jusqu’à son diagnostic correct. Les raisons en sont l’hétérogénéité de ses formes, la nécessité d’une confirmation histologique, la pénurie d’équipements spécialisés et les croyances erronées de certains cardiologues selon lesquelles il s’agit d’une maladie rare sans options de traitement.2,3
Cependant, ces aspects sont en train de changer. Le diagnostic a des implications sur la prise en charge des patients. Des thérapies spécifiques ont été développées qui peuvent retarder ou stabiliser le dépôt et qui sont plus efficaces dans les premiers stades. Un diagnostic précoce est donc crucial. Cette revue décrit les avancées récentes significatives dans le diagnostic et le traitement de l’ATTR, offrant ainsi de l’espoir aux patients atteints de cette maladie.
TRANSTHYRÉTINE AMYLOIDOSE CARDIQUE
La transthyrétine est une protéine plasmatique tétramérique responsable du transport de la thyroxine et des protéines liées au rétinol. Elle est principalement synthétisée dans le foie et secondairement dans les plexus choroïdes et l’épithélium pigmentaire rétinien.4
La transthyrétine a tendance à se dissocier en dimères et monomères, qui s’assemblent mal en fibrilles et subissent un dépôt. Des mutations ponctuelles ou l’effet de l’âge peuvent augmenter cette tendance, donnant lieu aux 2 formes cliniques de l’ATTR : mutante (ATTRm) et sauvage (ATTRwt).
AMYLOIDOSE DE TRANSTHYRÉTINE MUTANTE
Plus de 120 mutations sont actuellement connues pour causer l’ATTRm. Ces mutations présentent un mode d’hérédité autosomique dominant, avec une pénétrance variable4. En raison de sa grande diversité géographique, il est difficile d’établir la prévalence de l’ATTR, mais elle est considérée comme une maladie rare dont la prévalence est inférieure à 1/100 000 habitants2 (tableau 1).
Principales caractéristiques cliniques et diagnostiques de la transthyrétine cardiaque mutante et sauvage.Type Transthyrétine Amyloïdose Cardiaque
| ATTRwt | ATTRm | |
|---|---|---|
| Prévalence | Inconnue. Apparemment très fréquente | |
| Étude génétique | Absence de mutations dans la TTR | Mutation dans la TTR |
| Age typique à la présentation | > 60 ans | Variable selon la mutation causale |
| Sexe | Dominance masculine. 80% des patients | Dominance masculine, avec un phénotype plus agressif |
| Manifestations extracardiaques | – Syndrome du canal carpien (33%-49%) – Sténose spinale lombaire – Rupture traumatique du tendon du biceps (32%) |
– Polyneuropathie sensori-motrice bilatérale ascendante – Dysautonomie : hypotension orthostatique, diarrhée-constipation, dysfonctionnement érectile – Atteinte oculaire : glaucome, dépôt intravitréen, pupilles festonnées |
| Influence cardiaque | Constante | Variable selon la mutation causale |
| Débit cardiaque | – Insuffisance cardiaque (53%-86%) – Troubles de la conduction – FA (43%-67%) – AoS dégénératif |
– Troubles de la conduction – Insuffisance cardiaque – FA peu fréquente (10%) |
| Techniques de diagnostic | ||
| ECG | – Modèle de pseudo-infarctus (63%-66%) – Basse tension (22%-33%) – LVH de Sokolow (6%-13%) |
– Modèle de pseudo-infarctus (18%-69%) – Basse tension (2%-25%) – LVH de Sokolow (3%-8%) |
| ECHO | – Hypertrophie modérée-sévère – Dépression légère-modérée de la FEVG (30%) |
– Hypertrophie modérée – FEVG, typiquement préservée |
| IRM cardiaque | – Rehaussement tardif – T1 et EV natifs élevés |
|
| Scintigraphie au 99mTc DPD | – Grade 2-3 | – Grade 0 : porteurs asymptomatiques – Grade 1 : atteinte cardiaque initiale – Grade 2-3 : atteinte cardiaque importante |
AF, fibrillation auriculaire ; AoS, sténose aortique ; ATTRm, amyloïdose à transthyrétine mutante ; ATTRwt, amyloïdose à transthyrétine de type sauvage ; ECG, électrocardiogramme ; ECO, échocardiogramme ; VE, volume extracellulaire ; FEVG, fraction d’éjection du ventricule gauche ; HVG, hypertrophie ventriculaire gauche ; TTR, transthyrétine.
Les premières mutations de la TTR ont été rapportées comme une polyneuropathie amyloïde familiale (ou maladie d’Andrade), et par conséquent l’ATTRm a jusqu’à récemment été considérée comme une maladie neurologique. Cependant, des découvertes récentes montrent une atteinte cardiaque dans plus de la moitié des cas.3
Il existe une forte corrélation génotype-phénotype, les mutations étant associées à une maladie purement neurologique ou à une maladie purement cardiaque.3
Les mutations sont associées à une maladie purement neurologique ou à une maladie purement cardiaque. Cependant, la division de l’ATTRm en maladie cardiaque ou neurologique peut être une simplification excessive, car il existe un chevauchement considérable entre les 2 formes cliniques du spectre de la maladie.
La mutation Val30Met (maintenant connue sous le nom de Val50Met après l’ajout de 20 positions au nom de la mutation traditionnelle dans l’ATTRm) est la mutation la plus fréquente dans le monde et est endémique au Portugal, au Japon et en Suède. Son incidence estimée au Portugal est de 1 pour 538 habitants.2 Majorque (Espagne) et Valverde del Camino (Huelva, Espagne) sont également considérées comme des zones où l’ATTRm est endémique. La prévalence estimée à Majorque chez les patients symptomatiques est de 3/100 000 habitants.5
La mutation Val30Met provoque une affection principalement neurologique avec une polyneuropathie sensori-motrice symétrique, qui débute dans les membres inférieurs et suit un schéma ascendant. Elle peut être associée à une dysautonomie avec hypotension orthostatique, dysfonctionnement érectile, incontinence urinaire et symptômes gastro-intestinaux. Elle débute généralement à la fin de la deuxième ou de la troisième décennie de vie, et jusqu’à 43 % des patients présentent une atteinte cardiaque qui est une cause fréquente de décès4 (tableau 1).
La mutation Val122Ile (p. Val142Ile), qui est présente chez 3 % à 4 % de la population noire nord-américaine, revêt une importance particulière3. Bien que sa pénétrance soit incomplète,3 cette mutation a été associée à un risque accru de 47% de développer une insuffisance cardiaque (IC).6 Une étude récente a montré que l’amylose Val122Ile était la quatrième cause la plus fréquente d’IC dans la population afro-caribéenne britannique.7 Bien que jusqu’à 30% des patients présentant cette mutation puissent présenter des caractéristiques de neuropathie légère,6 le phénotype clinique est généralement similaire à celui de l’ATTRwt.4 La mutation Val122Ile ne doit pas être considérée comme une mutation exclusive à la population noire, car elle peut également être présente dans la population blanche. Par exemple, nous avons identifié cette mutation dans 4 familles espagnoles blanches sans ascendance noire.
WILD-TYPE TRANSTHYRETIN AMYLOIDOSIS
L’amylose à transthyrétine de type sauvage a été décrite pour la première fois en 1876. Elle était autrefois appelée amylose sénile, mais son diagnostic chez des patients âgés de 40 à 60 ans a rendu ce terme obsolète. Il est intéressant de noter que le plus ancien cas connu de cette mutation a été trouvé chez un patient américain de 47 ans.8
La prévalence exacte de l’ATTRwt reste inconnue. Cependant, des études suggèrent qu’elle est sous-diagnostiquée et qu’elle pourrait être la forme la plus fréquente d’amylose cardiaque.2,3 Les résultats suivants soutiennent cette hypothèse:
- –
Chez les patients âgés de plus de 80 ans, la prévalence du dépôt de TTR est de 25% à l’autopsie.3
- –
Chez les patients atteints d’HF avec fraction d’éjection préservée (HFpEF), le dépôt modéré-sévère de TTR est de 5% à l’autopsie.9
- –
Chez les patients âgés de plus de 60 ans admis pour une HFpEF et une hypertrophie ventriculaire gauche (LVH) ≥ 12mm, notre groupe a récemment trouvé une prévalence de 13%.10
Contrairement à l’ATTRm, l’ATTRwt est une maladie sporadique qui débute généralement après l’âge de 70 ans4. Elle se retrouve principalement chez les hommes, et les séries publiées ont rapporté des taux allant de 89% à 98%.11,12 Cependant, dans une série récente de patients diagnostiqués avec ATTRwt dans 2 hôpitaux (Madrid, Espagne et Bologne, Italie), notre groupe a constaté que 20% étaient des femmes. En outre, d’autres études autopsiques ont également suggéré que l’ATTRwt chez les femmes pourrait être plus répandu que ce qui avait été rapporté précédemment. Par conséquent, le sexe féminin ne devrait pas diminuer la suspicion clinique d’ATTRwt (tableau 1).13
Les résultats d’autopsie montrent que le dépôt de TTR est dispersé dans différents organes dans l’ATTRwt. Cependant, le dépôt est beaucoup plus important dans le cœur en raison du tropisme cardiaque de la TTR, et l’atteinte cardiaque est la principale manifestation clinique.4 Les patients peuvent présenter des symptômes de dépôt extracardiaque de TTR tels qu’une sténose du canal lombaire, une rupture atraumatique du tendon du biceps ou « signe de Popeye », et un syndrome du canal carpien (SCC)3 (Figure 1). Toutes ces caractéristiques peuvent aider à orienter et à établir rapidement le diagnostic. Le SCC peut accompagner d’autres sous-types d’amylose, mais il est plus fréquent dans l’ATTRwt. Le dépôt peut précéder de plusieurs années les manifestations cardiaques.6 Il peut être utilisé comme indication chez les patients âgés atteints d’HVG, en particulier s’ils présentent un SCC bilatéral non associé à des activités professionnelles spécifiques et s’ils sont en classe fonctionnelle ≥ II de la New York Heart Association (données non publiées).
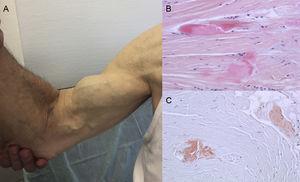
Signes et symptômes de l’amyloïdose à transthyrétine. A : rupture non traumatique du tendon du biceps droit (« signe de Popeye »). B et C : coloration à l’hématoxyline-éosine (B) et au rouge Congo (C), toutes deux ×200, d’un échantillon de ligament carpien montrant des faisceaux de collagène denses avec du matériel non cellulaire. Avec l’aimable autorisation du Dr Clara Salas Antón.
DIAGNOSTIC DE L’AMYLOIDOSE DE LA TRANSTHYRÉTINE Présentation clinique
L’amyloïde peut infiltrer n’importe quelle structure cardiaque1. Typiquement, le dépôt augmente l’épaisseur de la paroi ventriculaire, ce qui entraîne une diminution progressive de la distensibilité conduisant à une dysfonction diastolique sévère. L’ATTR a donc traditionnellement été incluse comme une cause de cardiomyopathie restrictive.
Cependant, le spectre clinique de l’ATTR est beaucoup plus large et plus hétérogène. Le symptôme le plus courant de l’ATTR est l’HF. Comme mentionné, une étude publiée par notre groupe en 2015 a suggéré qu’un protocole basé sur la scintigraphie au 99mTc-3,3-diphosphono-1,2-propanodicarboxylique (99mTc-DPD) peut être utile pour le diagnostic de l’ATTRwt chez une proportion significative (13 %) de patients âgés de plus de 60 ans admis pour une FHpEF.10 Sur la base de ce résultat, la scintigraphie au 99mTc-DPD a été incluse dans les lignes directrices européennes de 2016 sur l’HF comme un outil utile pour l’identification des patients atteints d’ATTR14. Cependant, l’ATTR ne doit pas être suspectée exclusivement chez les patients atteints d’HFpEF car, à mesure que le dépôt amyloïde progresse, la fonction contractile s’aggrave et, par conséquent, l’ATTR peut être associée à différents degrés de dysfonctionnement systolique.
L’amyloïdose à transthyrétine est une phénocopie de la cardiomyopathie hypertrophique (CMH) et peut être confondue avec elle. Une étude française multicentrique récente a rapporté que 5% des patients atteints de HCM ont une ATTRm.15 Cependant, nos résultats ne correspondent pas à ce taux élevé, qui pourrait être lié à la grande population noire en France.
Les anomalies de la conduction cardiaque peuvent être la première manifestation de l’ATTR. L’infiltration amyloïde du sinus et des ganglions auriculo-ventriculaires1 peut indiquer la nécessité d’implanter un stimulateur cardiaque (tableau 1). L’étude mentionnée précédemment, menée en Espagne et en Italie, a révélé que les troubles de la conduction étaient la première manifestation de l’ATTRwt chez 7 % des patients atteints de cette maladie13
Les arythmies auriculaires sont également très fréquentes chez les patients atteints d’ATTRwt13 (figure 2A), et la première manifestation de la maladie peut être un accident vasculaire cérébral4. En fait, le groupe de la Mayo Clinic a récemment suggéré que l’ATTRwt devrait être exclu dans le cadre d’un diagnostic de fibrillation auriculaire (FA) non valvulaire chez les patients âgés.8 En revanche, la FA est beaucoup moins fréquente chez les patients atteints d’ATTRm (tableau 1).
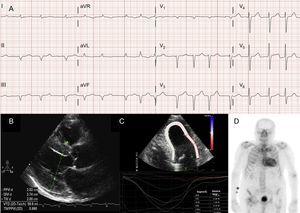
Techniques de diagnostic dans l’amylose cardiaque à transthyrétine (ATTR). A : électrocardiogramme d’un patient atteint d’amylose transthyrétine de type sauvage (ATTRwt), montrant une fibrillation auriculaire et un motif de pseudo-infarctus dans les dérivations inférieures. B : échocardiogramme d’un patient atteint d’amylose mutante à transthyrétine avec mutation Val30Met, présentant une hypertrophie concentrique marquée du ventricule gauche et un léger épanchement péricardique. C : souche régionale longitudinale d’un patient atteint d’ATTRwt, montrant des valeurs préservées dans le segment apical et des valeurs déprimées dans les segments basal et médio-ventriculaire. D, scintigraphie au 99mTc-DPD (acide 99mTc-3,3-diphosphono-1,2-propanodicarboxylique) d’un patient atteint d’ATTRwt, montrant une captation biventriculaire supérieure à la captation osseuse, correspondant au grade 3 de Perugini.
Enfin, nous notons que l’ATTR et la sténose aortique dégénérative peuvent coexister chez le même patient. En 2016, plusieurs études ont attiré l’attention sur cette possibilité, et une étude prospective a rapporté que l’ATTRwt avait une prévalence de 6 % chez les patients de plus de 65 ans qui ont subi un remplacement de la valve aortique16. Cette étude a suggéré que les patients présentant les deux entités avaient un pronostic postopératoire bien plus mauvais que ceux ne présentant pas d’ATTRwt (mortalité de 50 % contre 6,9 % après un suivi médian de 2,3 ans).16 Une autre étude récente avec une scintigraphie au 99mTc-DPD chez 43 patients présentant une sténose aortique à faible débit/faible gradient a identifié 5 patients présentant une ATTRwt (prévalence de 12 %). 17 Les patients atteints de sténose aortique sévère et d’ATTRwt partagent le même profil démographique, et le traitement approprié pour les patients atteints des deux maladies reste à déterminer.
Utilité des techniques de diagnostic
Le diagnostic de l’ATTR est un défi dans la pratique clinique quotidienne. Bien que l’électrocardiographie et l’échocardiographie jouent un rôle dans le diagnostic, de nouvelles techniques non invasives ont acquis un rôle clé dans l’évaluation des patients atteints d’ATTR.
Électrocardiogramme
L’association entre la tension basse et l’amylose cardiaque a longtemps été considérée comme indiscutable3. Les critères les plus utilisés dans la pratique clinique sont l’amplitude du QRS 1. Bien que de faibles tensions électrocardiographiques dans le cadre d’une LVH doivent établir la suspicion, la prévalence dans une série contemporaine d’ATTR n’était que de 20 à 25 %.3,4,13 La prévalence varie également en fonction des critères appliqués. Par exemple, l’utilisation du critère de Sokolow (onde S dans la sonde V1 + onde R dans la sonde V5 ou V6
1,5 mV) peut augmenter la prévalence calculée entre 46 % et 58 %.13 Le rapport entre l’épaisseur de la paroi ventriculaire gauche et le voltage QRS total a été recommandé pour mieux évaluer les disparités entre les résultats des 2 techniques.2,3 Cependant, jusqu’à 20 % des patients atteints d’ATTR peuvent répondre aux critères électrocardiographiques de l’HVG.2,3
Dans la plupart des séries de patients atteints d’amylose cardiaque, le schéma pseudo-infarctus est la découverte électrocardiographique la plus courante2,3,13 (Figure 2A). En raison de l’implication possible du système de conduction, les blocs de branche complets ou incomplets sont également fréquents.3
Échocardiographie
Bien que l’échocardiographie soit la pierre angulaire du diagnostic initial de l’ATTR, aucun résultat n’est spécifique.3 L’amylose à transthyrétine a été typiquement associée à un ventricule gauche normal ou petit avec une hypertrophie concentrique.3 Le 10e Symposium international sur l’amyloïde et l’amylose, qui s’est tenu en 2004, a établi le critère échocardiographique d’une cardiopathie due à l’AL en l’absence d’autres causes d’HVG comme étant la présence d’une HVG avec un seuil de 12 mm pour l’épaisseur de la paroi du septum interventriculaire4. Ce critère a ensuite été extrapolé à d’autres formes d’amylose (Figure 2B), ce qui a conféré un haut degré de spécificité mais une faible sensibilité.
Bien que l’HVG concentrique ait été classiquement décrite, les séries actuelles suggèrent qu’environ 20% ont une HVG asymétrique.13
Malgré l’association classique entre une fraction d’éjection ventriculaire gauche (FEVG) normale ou légèrement diminuée et l’amylose cardiaque,2 la fourchette de FEVG est très variable.8 Dans une étude récente menée à la Mayo Clinic, une FEVG de 8 alors que dans notre série, une FEVG de 13 En outre, l’utilisation de la FEVG dans l’évaluation de la fonction systolique dans l’amylose cardiaque est limitée, car des valeurs légèrement déprimées sont déjà indicatives d’une maladie cardiaque pertinente. Cette limitation peut être surmontée par l’utilisation des vitesses Doppler tissulaires, de l’imagerie de déformation et de la fraction de contraction myocardique, qui ont été proposées comme des indices plus appropriés pour évaluer la fonction cardiaque.2
Les autres signes échocardiographiques classiques sont l’hypertrophie du ventricule droit, la dilatation bi-auriculaire, un léger épanchement péricardique, l’épaississement de la valve auriculo-ventriculaire, l’épaississement de la paroi du septum auriculaire et l’aspect granuleux scintillant du myocarde.3,6 Cependant, comme certaines de ces caractéristiques ont été observées dans une série hautement sélectionnée de patients à des stades avancés de la maladie, il n’est pas nécessaire qu’elles soient toutes présentes pour établir une suspicion.1
L’imagerie de déformation régionale est une technique très utile pour le diagnostic précoce des patients atteints d’ATTR. Chez les patients atteints d’ATTR, le strain longitudinal est déprimé dans les segments basaux et médio-ventriculaires mais est préservé dans les segments apicaux18 (Figure 2C). Ce schéma typique peut être utile dans le diagnostic différentiel de l’ATTR par rapport à d’autres maladies cardiaques.4
Biomarqueurs
On dispose de moins de données sur le rôle du prohormone N-terminale du propeptide natriurétique cérébral (NT-proBNP) et de la troponine dans l’ATTR que dans l’AL.4 Les taux de NT-proBNP dans l’ATTR sont typiquement plus faibles que dans la LA,4 ce qui reflète 2 mécanismes physiopathologiques différents : toxicité directe de la chaîne légère dans la LA vs dommages tissulaires induits par les protofibrilles dans l’ATTR.
Récemment, le groupe de la Mayo Clinic a proposé un système de stratification similaire à celui en vigueur pour la LA. Dans une cohorte de 360 patients atteints d’ATTRwt, les deux biomarqueurs se sont révélés être des facteurs prédictifs de mortalité. Les patients de stade III (NT-proBNP > 3000 pg/mL et troponine T > 0,05 ng/mL) ont eu une survie médiane de 20 mois, alors que les patients de stade I et II ont eu une survie médiane de 66 mois et 40 mois (aucun biomarqueur ou seulement 1 biomarqueur au-dessus des seuils établis, respectivement).
Imagerie par résonance magnétique cardiaque
L’imagerie par résonance magnétique cardiaque (IRMC) peut être utilisée pour obtenir des informations structurelles et fonctionnelles et caractériser la composition du tissu myocardique3. L’IRMC est essentielle dans l’identification précoce de l’ATTR, et dans son diagnostic différentiel avec d’autres maladies cardiaques.
La caractérisation des tissus par l’IRMC est basée sur les caractéristiques suivantes :
- –
Renforcement tardif : Un motif global sous-endocardique est pratiquement pathognomonique de l’amylose cardiaque, mais il n’est présent que chez environ un quart des patients. D’autres schémas, tels que le schéma transmural (le plus fréquent) ou le patch, sont également compatibles (Figure 3). Malgré sa sensibilité et sa spécificité élevées, il faut tenir compte de l’absence possible de rehaussement tardif (15% des patients) et, selon notre expérience, d’un pourcentage non négligeable de faux négatifs pour des raisons techniques.3 Le schéma de rehaussement transmural est associé à un pronostic plus défavorable et constitue un facteur prédictif indépendant de mortalité.19
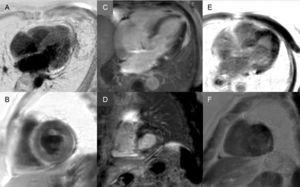 Figure 3.
Figure 3.Diversité des séquences de rehaussement tardif par imagerie par résonance magnétique cardiaque dans l’amyloïdose à transthyrétine. A et B : séquences de rehaussement tardif, plan de la 4-chambre et axe court au niveau moyen, respectivement, d’un patient atteint d’amylose à transthyrétine mutante (ATTRm), montrant un dépôt de gadolinium transmural pathologique diffus. C et D : séquences de rehaussement tardif, respectivement au niveau de la chambre 4 et de l’axe court au niveau basal, de patients atteints d’ATTRm, montrant un dépôt de gadolinium pathologique avec un motif en forme de patch, avec une zone focale basale inférieure inferoseptale et inferolatérale. E et F, séquences de rehaussement tardif, plan de 4 chambres et axe court au niveau apical, respectivement, de patients atteints d’ATTRm, montrant un dépôt pathologique transmural étendu, sauf dans les segments basal et antérolatéral moyen. Courtoisie du Dr Jesús González Mirelis.
(0.15MB). - –
Temps T1 longs : La cartographie T1 est une technique dans laquelle un signal myocardique quantitatif est mesuré avant (T1 natif) ou après l’administration d’un contraste. Les temps T1 natifs sont très longs dans l’amylose cardiaque.3 La cartographie T1 ne nécessite pas l’administration de contraste et peut donc être utilisée dans l’insuffisance rénale. Les temps T1 peuvent même être anormaux avant l’observation d’une LVH.3 Les temps T1 sont plus longs dans l’ATTR que dans la HCM et les contrôles (1097ms ± 43 ms vs 1026ms ± 64 ms vs 9,67ms ± 34ms, respectivement ; P
ms ± 68 ms ; P = 0,01).20
L’administration de contraste peut être utilisée pour calculer le volume extracellulaire (ECV) et évaluer les augmentations de l’espace extracellulaire. Les valeurs du VCE dans l’amylose cardiaque sont plus élevées que dans d’autres maladies cardiaques, sauf dans les zones d’infarctus du myocarde.21 En 2016, notre groupe, en collaboration avec d’autres centres nationaux, a rapporté que la quantification du VCE peut identifier l’atteinte cardiaque dans l’ATTRm et, pour la première fois, l’a corrélée avec le degré d’atteinte neurologique, soutenant l’utilisation de cette technique dans le diagnostic précoce et le suivi de l’ATTRm22.
Les techniques de cartographie T1 quantitative et de calcul de la VCE peuvent être très utiles dans l’ATTR pour le diagnostic précoce, le suivi clinique et l’évaluation de la réponse au traitement (figure 4).
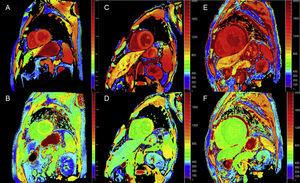
Cartographie T1, avant et après contraste, avec look-locker modifié inversion-récupération (MOLLI) en imagerie par résonance magnétique cardiaque 3T chez un témoin sain, un patient atteint d’amyloïdose à transthyrétine et un patient atteint d’amyloïdose primaire à chaîne légère. A et B : cartographie T1 native et volume extracellulaire (VE), respectivement, chez un témoin sain, montrant des valeurs normales (VE = 0,214). C et D : cartographie T1 native et volume extracellulaire (VE), respectivement, chez un patient atteint d’amylose mutante à transthyrétine, présentant des lésions neurologiques et un début d’atteinte cardiaque, avec un T1 natif élevé et un VE légèrement élevé (0,361). E et F : cartographie T1 native et EV, respectivement, chez un patient atteint d’amylose cardiaque à transthyrétine de type sauvage, T1 native élevée et EV très élevée (0,626), reflétant une infiltration amyloïde massive. Courtoisie du Dr Jesús González Mirelis.
Scintigraphie cardiaque
Dans les années 1980, l’observation de la captation cardiaque de plusieurs traceurs diphosphonates osseux a été histologiquement corrélée à la présence d’une amylose cardiaque23. Le mécanisme de la captation n’est pas bien caractérisé, mais il pourrait être lié à la teneur en calcium des dépôts amyloïdes.
Une étude précoce du groupe de Bologne utilisant le 99mTc-DPD a trouvé une captation cardiaque chez 15 patients atteints d’ATTR et son absence chez 10 patients atteints d’AL, en utilisant un score basé sur la captation biventriculaire égale ou supérieure à la captation osseuse (score de Perugini)24 (Figure 2D). Des résultats similaires ont été rapportés par la suite par notre groupe et par d’autres.25 Une captation légère (score 1) et une captation modérée (score 2) peuvent être trouvées chez 30% et 10% des patients atteints de la maladie d’Alzheimer, respectivement.24
Etant donné sa sensibilité et sa spécificité élevées, cette technique est extrêmement utile pour établir un diagnostic d’ATTR et peut montrer une atteinte cardiaque même lorsque les résultats de l’échocardiographie et de l’IRM sont encore normaux. En fait, après une scintigraphie pour des indications oncologiques ou rhumatologiques, les découvertes fortuites d’ATTR ne sont pas rares.26
Le Tc-DPD n’est pas disponible aux États-Unis, mais des résultats similaires ont été rapportés en utilisant l’imagerie au 99mTc-PYP (pyrophosphate).27
D’autres radiotraceurs sont actuellement à l’étude. Par exemple, le 18F-florbetapir, qui a déjà été approuvé pour l’imagerie de la bêta-amyloïde cérébrale,4 a été étudié chez des patients atteints de AL et d’ATTR. Les résultats montrent que le 18F-florbétapir peut détecter les dépôts myocardiques d’AL et d’ATTR.28 Bien que les données disponibles aient été obtenues dans le cadre d’études de cas29 et que le coût élevé de ce radiotraceur limite son utilisation, plusieurs études sont en cours sur l’avantage potentiel de son utilisation par rapport à celle du Tc-DPD comme technique de dépistage des 2 types d’amylose les plus courants.
Diagnostic invasif
Le diagnostic définitif de l’ATTR repose sur la démonstration histologique de fibrilles amyloïdes. Bien qu’il puisse y avoir un dépôt extracardiaque, la probabilité de démontrer l’amyloïde par histologie varie selon l’organe.2 Il existe peu d’études sur la rentabilité de la biopsie extracardiaque (par exemple, graisse abdominale, gingivale, glande salivaire, gastro-intestinale) dans l’ATTR, qui est plus grande dans l’ATTRm que dans l’ATTRwt. Cependant, une biopsie négative d’un organe cliniquement non affecté n’exclut pas un diagnostic d’ATTR.4
Comme dans l’ATTRwt, la biopsie endomyocardique est indiquée chez les patients sans atteinte extracardiaque ou avec une maladie cardiaque seule.3,4 La biopsie endomyocardique est une procédure à faible risque (surtout dans les centres expérimentés) et les erreurs d’échantillonnage sont peu probables.6
Après confirmation histologique de l’amylose, qui peut parfois nécessiter une interprétation par un personnel qualifié,6 une classification correcte du sous-type est cruciale.4 Actuellement, la classification dépend d’une combinaison d’immunohistochimie, d’analyse génétique et de protéomique :
- –
L’immunohistochimie repose sur l’utilisation d’anticorps spécifiques contre des protéines amyloïdes connues. Bien que les résultats de cette technique soient généralement définitifs, elle est moins sensible dans la reconnaissance des chaînes légères.4
- –
Cette limitation peut être surmontée par l’utilisation de la spectrométrie de masse, qui fournit des résultats définitifs et constitue le critère standard dans la confirmation du sous-type amyloïde.2 Bien que cette technique ne soit disponible que dans les centres spécialisés, elle est particulièrement utile dans les cas non concluants ou dans les cas qui sont positifs pour plusieurs anticorps à l’immunohistochimie, ce qui, dans notre expérience, se produit dans environ 20 à 30% des cas. 4
- –
Parce que les techniques cliniques ou histologiques ne permettent pas de distinguer l’ATTRm de l’ATTRwt, des études génétiques sont recommandées dans tous les cas d’ATTR. La découverte d’une mutation causale peut être importante pour offrir un conseil génétique et un suivi aux porteurs asymptomatiques, 4,30 qui pourraient bénéficier des thérapies à venir qui retardent ou même préviennent l’apparition de la maladie31.
Diagnostic non invasif
Jusqu’à récemment, les études histologiques étaient considérées comme essentielles dans le diagnostic de l’ATTR.3 Cependant, pour faciliter le diagnostic, une étude multicentrique internationale a proposé en 2016 un nouvel algorithme pour le diagnostic non invasif de l’ATTR.32
L’étude a analysé les résultats de 1217 patients. La présence de signes classiques d’amylose cardiaque à l’aide de techniques d’imagerie, une captation de Tc-DPD/PYP de grade 2 ou 3 à la scintigraphie et l’absence de protéine monoclonale avaient une spécificité et une valeur prédictive positive pour l’ATTR de 100 %32 (figure 5).
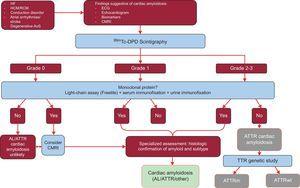
Algorithme diagnostique pour les patients présentant une suspicion d’amylose cardiaque. Système de gradation de la scintigraphie au 99mTc-DPD : grade 0, pas de captation cardiaque ; grade 1, captation légèrement inférieure à la captation osseuse ; grade 2, captation modérée égale à la captation osseuse ; grade 3, captation sévère supérieure à la captation osseuse. ACV, accident vasculaire cérébral ; AL, amylose primaire à chaîne légère ; AoS, sténose aortique ; ATTR, amylose à transthyrétine ; ATTRm, amylose à transthyrétine mutante ; ATTRwt, amylose à transthyrétine de type sauvage ; CMRI, imagerie par résonance magnétique cardiaque ; ECG, électrocardiogramme ; HCM, cardiomyopathie hypertrophique ; HF, insuffisance cardiaque ; RCM, cardiomyopathie restrictive ; TTR, transthyrétine.
Une caractéristique clé de cet algorithme est l’absence d’une protéine monoclonale qui pourrait provoquer une AL sur le dosage de la chaîne sérique (Freelite, The Binding Site, UK) et sur l’électrophorèse par immunofixation du sang et de l’urine. La présence d’une protéine monoclonale est une indication pour une biopsie endomyocardique afin de distinguer l’ATTR de la AL.32 Jusqu’à 5 % de la population âgée de plus de 65 ans présentent une gammapathie monoclonale de signification indéterminée.2 Chez les personnes âgées, une augmentation modérée des chaînes légères circulantes ne doit pas conduire directement à un diagnostic de AL. Il a été signalé que jusqu’à 10 % des patients âgés présentant une ATTRwt et une gammapathie monoclonale de signification indéterminée dans les centres de référence avaient auparavant reçu un diagnostic erroné de LA.3,33 Un diagnostic correct est nécessaire pour éviter une chimiothérapie inappropriée. Il est intéressant de noter que notre hôpital a documenté 2 cas de patients atteints de myélome multiple et d’ATTRwt concomitante sur la spectrométrie de masse.
Traitement de l’AMYLOIDOSE CARDIQUE TRANSTHYRETINE
Le traitement des patients atteints d’ATTR a 2 objectifs : fournir un soutien médical et, si possible, arrêter ou retarder le dépôt amyloïde par l’utilisation de traitements spécifiques.
Traitement médical
Les sections suivantes décrivent les soins cardiaques de soutien pour les patients atteints d’ATTR.
Gestion de l’insuffisance cardiaque
L’euvolémie doit être maintenue chez les patients atteints d’amylose cardiaque. Les mesures relatives au régime alimentaire et au mode de vie sont très importantes. Les diurétiques sont essentiels au traitement de l’HF dans l’ATTR. Cependant, étant donné que l’utilisation excessive de diurétiques peut entraîner une hypotension (fréquemment due à un dysfonctionnement autonome) et aggraver la situation clinique, en particulier dans l’ATTRm, une extrême prudence doit être exercée dans sa gestion.
Dans le traitement de l’HF dans l’ATTR, il faut tenir compte du fait que la dysfonction diastolique altérée et le volume d’attaque réduit entraînent une tachycardie compensatoire pour maintenir le débit cardiaque. Par conséquent, les bêta-bloquants doivent être utilisés avec précaution et individualisés pour chaque patient. La pratique courante consiste à les supprimer en l’absence de difficultés à contrôler la fréquence cardiaque. Cette approche est encore plus importante, si possible, dans l’ATTRwt en raison de la présence fréquente de troubles de la conduction.6 Les antagonistes calciques et la digoxine peuvent se lier aux fibrilles amyloïdes et sont donc contre-indiqués dans l’ATTR en raison du risque de toxicité, même à des doses thérapeutiques6.
Contrairement à l’HF avec dysfonction systolique due à d’autres étiologies, il n’existe aucune preuve en faveur d’un bénéfice pronostique dû à l’utilisation de bêta-bloquants, d’inhibiteurs de l’enzyme de conversion de l’angiotensine ou d’antagonistes des récepteurs de l’angiotensine II dans l’amylose cardiaque. En fait, leur utilisation peut conduire à une aggravation clinique due à l’hypotension et à un faible débit : une publication récente a rapporté un pronostic plus mauvais dans l’ATTRm et un effet neutre dans l’ATTRwt.34
Gestion des arythmies auriculaires
La gestion de la FA dans l’ATTR est un défi. Le maintien à long terme du rythme sinusal est difficile. Cependant, une cardioversion électrique peut être tentée car elle peut conduire à une amélioration clinique.
Le risque thromboembolique chez les patients atteints d’ATTR est très élevé. En outre, l’infiltration amyloïde chronique peut entraîner une dysfonction auriculaire mécanique, qui peut être la cause sous-jacente du développement d’un thrombus auriculaire chez certains patients sans FA. Le traitement anticoagulant dans l’ATTR ne doit pas être basé sur le score CHADS2-VASC et doit être un traitement standard dans la FA. Les événements hémorragiques sont moins fréquents que dans la FA, c’est pourquoi certains hôpitaux recommandent un traitement anticoagulant chez les patients en rythme sinusal si la fonction auriculaire est médiocre d’après les vitesses du Doppler transmitral. Bien qu’il n’existe pas d’études comparatives sur l’efficacité des anticoagulants oraux directs par rapport aux antagonistes de la vitamine K, notre hôpital a administré des anticoagulants oraux directs à des patients sélectionnés.
Rôle des dispositifs
Les recommandations actuelles pour l’implantation d’un stimulateur cardiaque sont les mêmes dans l’ATTR et dans la population générale. Cependant, certains groupes favorisent l’implantation prophylactique, en particulier chez les patients atteints d’ATTRm et de troubles de la conduction.35 Nous ne sommes pas favorables à cette stratégie préventive et n’avons pas trouvé un taux si élevé de troubles de la conduction pour justifier une implantation prophylactique chez les patients atteints d’ATTRm.
Le rôle de l’utilisation d’un cardioverteur-défibrillateur implantable (ICD) dans l’ATTR n’est pas bien établi. Dans une petite série, l’implantation d’un DAI n’a pas amélioré de manière significative la survie, bien qu’elle ait eu un effet approprié chez plusieurs patients au cours des 2 premières années.36
Transplantation cardiaque
La transplantation cardiaque a joué un rôle mineur dans l’ATTR car l’ATTRm peut impliquer divers organes et l’ATTRwt affecte généralement les patients âgés. Cependant, l’absence d’atteinte extracardiaque chez les patients atteints d’ATTRwt fait d’eux de bons candidats pour cette procédure. La littérature fournit des exemples de transplantation réussie chez des patients de moins de 70 ans atteints d’ATTRwt ou d’ATTRm et d’une atteinte cardiaque prédominante.4
Traitement spécifique de l’amylose cardiaque à transthyrétine
À l’heure actuelle, il n’existe pas de thérapie approuvée pour le traitement spécifique de l’amylose cardiaque à ATTR, bien que la transplantation hépatique (TxH), seule ou en association avec la transplantation cardiaque, soit utilisée dans l’ATTRm depuis les années 1990 comme moyen d’éliminer la principale source de précurseurs de la TTR4.
Transplantation hépatique
Le registre mondial des transplantations de polyneuropathie amyloïde familiale37 a rapporté que plus de 2000 patients atteints d’ATTRm ont subi une TxH dans 20 pays.4 Les patients présentant la mutation Val30Met et un tableau clinique à prédominance neurologique ont un taux de survie de plus de 50% à 20 ans.3 Ces résultats prometteurs reposent sur une sélection stricte des patients en fonction de l’âge, du type de mutation et du stade de la maladie. L’indication la plus communément admise pour la TxH est la combinaison d’un jeune âge, de la mutation Val30Met et de stades précoces de la maladie.
Cependant, les principales limites de cette technique sont la pénurie de donneurs, la nécessité d’une immunosuppression chronique, l’âge avancé au moment de la présentation et les moins bons résultats obtenus chez les patients présentant des mutations autres que la mutation Val30Met.
En outre, la suppression théorique de la production de la protéine mutée est contrecarrée par le dépôt progressif de TTR native postimplantation,4,6 dont le mécanisme n’est pas complètement compris. En fait, le dépôt de TTR cardiaque après la TxH affecte la morbidité et la mortalité.
La nécessité de mieux comprendre la pathogenèse de l’ATTR et les limites de la TxH a stimulé le développement de plusieurs médicaments.
Ces nouveaux composés agissent à différents points de la cascade d’amyloïdogénèse de la TTR (figure 6). Le traitement consistera toujours à réduire la protéine précurseur, même si éviter les dépôts et éliminer les dépôts existants sera tout aussi important. Par conséquent, nous pensons qu’à l’avenir, l’approche de cette maladie prendra la forme d’un traitement combiné.
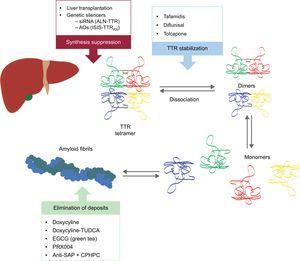
Traitements spécifiques dans l’amylose cardiaque à transthyrétine et principales cibles. AntiSAP + CPHPC, antisérum composant P amyloïde + acide (R)-1–6-oxo-hexanoyl]pyrrolidine-2-carboxylique ; AOs, oligonucléotides antisens ; EGCG, épigallocatéchine-3 gallate ; siRNA, petit ARN interférent ; TTR, transthyrétine ; TUDCA, acide tauroursodésoxycholique.
Suppression de la synthèse de la transthyrétine
Deux axes de recherche sont en cours sur l’inhibition de l’expression de la TTR hépatique : l’utilisation de petits ARN interférents (siRNA) et l’utilisation de médicaments à base d’oligonucléotides antisens (AO).
- –
Les siRNA sont des molécules d’ARN double brin qui réduisent au silence les séquences d’ARN messager en s’y liant spécifiquement, empêchant ainsi la formation de protéines. On a constaté que le patisiran (ALN-TTR02) réduit la production de TTR de 80 %.38 Chez les patients atteints d’ATTRm, la réduction de la TTR était de 87 %.39 Un essai de phase 2 a donné des résultats prometteurs, montrant des paramètres échocardiographiques, fonctionnels et analytiques stables à 12 mois et 24 mois.40 Les résultats de l’étude neurologique de phase 3 chez les patients atteints d’ATTRm et une sous-analyse des patients présentant une atteinte cardiaque sont attendus en 2017 (tableau 2). Un autre médicament, le revusiran (ALN-TTR01), est administré par voie sous-cutanée et diffère du patisiran par les nanoparticules lipidiques qui encapsulent le siRNA. Ce médicament a fait l’objet d’un essai clinique de phase III chez des patients atteints d’ATTRm et présentant une maladie cardiaque. L’étude a été interrompue l’année dernière en raison d’une augmentation inattendue de la mortalité dans le groupe traité (tableau 2).
Tableau 2.Principaux essais cliniques en cours dans l’amylose cardiaque à transthyrétine
Mécanisme d’action Composé Essai Conception. Patients (N) et sous-type d’ATTR Intervention Principaux critères d’évaluation Situation/résultats Suppression de la synthèse de la TTR Patisiran (ALN-…TTR02) NCT01961921 Etude de phase II, multicentrique 27
ATTRm (11 atteintes cardiaques)Patisiran 0.30 mg/kg IV toutes les 3 semaines pendant 2 ans Sécurité à long terme. Critères d’évaluation secondaires : effet sur les troubles neurologiques et les paramètres cardiaques Médicament bien toléré, avec un profil de sécurité similaire dans le phénotype neurologique et cardiaque
Troponine I, NT-proBNP et les données échocardiographiques sont restées stables à 12 mo et 24 moNCT01960348 (APOLLO) Phase III, randomisée, en double aveugle, contrôlée par placebo, multicentrique 225
ATTRm avec atteinte neurologiquePatisiran en perfusion IV vs placebo 2 :1 Changements dans le mNIS+7 Attendu en novembre 2017
Sous-analyse des patients avec une atteinte cardiaque préditeNCT02510261 Étude d’extension d’APOLLO Patisiran en perfusion IV vs placebo 2 :1 pendant 52 semaines Sécurité et effets indésirables à long terme En cours Revusiran (ALN-TTR01) NCT02319005 (ENDEAVOUR) Phase III, randomisée, en double aveugle, contrôlée par placebo 206
ATTRm avec atteinte cardiaqueRevusiran 500mg 5 j, puis hebdomadaire pendant 2 ans contre placebo Changements dans le test de marche de 6 m et dans les valeurs plasmatiques de la TTR Annulation en raison de l’augmentation de la mortalité dans le bras revusiran ISIS-TTRRX NCT01737398 Phase II/III, randomisée, en double aveugle, contrôlée par placebo, multicentrique 172
ATTRm avec neuropathie ; 50% d’atteinte cardiaque concomitanteIS-TTRRX 300mg SC toutes les 12 h pendant 1 wk, puis hebdomadaire pendant 64 semaines vs placebo Changements dans le mNIS+7 et le questionnaire de qualité de vie Norfolk Attendu en septembre 2017
Des cas de thrombocytopénie sévère et d’hémorragie ont été rapportés
Analyse des paramètres échocardiographiques et du NT-.proBNP attendue chez les patients sans hypertension avec LVH > 12 mmTraitement de la cardiomyopathie TTR avec un oligonucléotide antisens spécifique de la TTR Phase II, ouverte, non randomisée 20
ATTRm avec atteinte cardiaque et ATTRwtIS-TTRRX 300mg SC toutes les 12 h/semaine Paramètres échocardiographiques et IRM cardiaque vs contrôles historiques Pas de détérioration de la déformation et diminution de la masse du VG d’environ 5%
6 patients ont terminé 12 mo ; 15 patients 6 mo ; 1 patient TxCNCT02627820 Phase II, ouverte, non randomisée 50
ATTRwtISIS-TTRRX 300mg SC toutes les 12 h pendant 1 wk, puis 1 wk pendant 18 wk Changements dans la souche mesurée par speckle tracking Annulé sans initier le recrutement de patients Etude de phase III avec ISIS-TTRRX pour le traitement de la cardiopathie amyloïde TTR Phase III, randomisée, en double aveugle, contrôlée par placebo, multicentrique 490
ATTRwt et ATTRm avec atteinte cardiaqueISIS-TTRRX 300mg SC toutes les 12 h pendant 1 sem, puis hebdomadaire pendant 16 sem avec placebo, puis hebdomadaire pendant 24 mo Décès, TxC, ou admission pour causes cardiovasculaires En attente Stabilisation de la TTR Tafamidis NCT01994889 Phase III, randomisée, en double aveugle, contrôlée par placebo, multicentrique 441
ATTRwt et ATTRm avec atteinte cardiaqueTafamidis 20mg ou 80mg par voie orale toutes les 24 h pendant 30 mo vs placebo Mortalité toutes causes et hospitalisations cardiovasculaires Toutes causes et hospitalisations cardiovasculaires .cause et hospitalisations cardiovasculaires Fin février 2018 NCT02791230 Extension Phase III NCT01994889 330
ATTRwt et ATTRm avec atteinte cardiaqueTafamidis 20 mg ou 80 mg par voie orale toutes les 24 h pendant 60 mo Toute-…mortalité toutes causes et incidence des effets indésirables Prévue en décembre 2021 NCT00935012 Phase II, essai ouvert sur l’efficacité et la sécurité 31
ATTRwt ou ATTRm p.Val122Ile avec atteinte cardiaqueTafamidis 20mg par voie orale Sécurité et efficacité En cours jusqu’en décembre 2021 Diflunisal NCT00294671 Phase III, randomisée, en double aveugle, contrôlée par placebo, multicentrique 130
ATTRm avec phénotype neurologique (50% avec atteinte cardiaque)Diflunisal 250mg par voie orale toutes les 12 h vs placebo sur 24 mo NIS+7 à 24 mo NIS+7 diflunisal vs placebo 16.3 (P Pas de réduction de l’épaisseur ou de la tension ventriculaire chez les patients avec atteinte cardiaque vs placebo Elimination des dépôts Doxycycline + TUDCA/UDCA NCT01171859 Phase II, ouverte, non randomisée, prospective 40
ATTR (25 ATTRm, 13 ATTRwt, et 2 transplantés hépatiques domino)Doxycycline 100 mg toutes les 12 h + TUDCA 250 mg toutes les 8 h pendant 12 mo, puis 6 mo sans traitement Augmentation 14 patients se sont retirés
Réactions cutanées indésirables, 16 patients
68% des 25 patients évaluables ont rempli le critère principal
Amélioration globale de la souche à 12 mo et aggravation après 6 mo sans traitementNCT01855360 Phase II, ouverte, non randomisée, prospective par rapport aux contrôles historiques 30
Amyloïdose ATTR cardiaque (27 ATTRwt et 3 ATTRm). Contrôles historiques, 14 patients ATTRwtDoxycycline 100 mg toutes les 12 h + TUDCA 250 mg toutes les 8 h pendant 18 mo Changements de la souche longitudinale tous les 6 mo 22 patients ont terminé l’étude et étaient évaluables
Détérioration plus importante de la souche chez les contrôles par rapport au groupe de traitement
Augmentation du NT-proBNP dans le groupe de traitement ; non mesuré chez les témoinsNCT01677286 Phase II, ouverte, non randomisée, prospective 25
Amyloïdose systémique (6 ATTRwt et 3 ATTRm)Doxycycline 100 mg toutes les 12 h pendant 12 mo Sécurité du médicament
Réponse des organes atteintsAugmentation du NT-proBNP et de la fonction rénale
Aucune amélioration des autres paramètres étudiés
60% des patients ont présenté des complications cutanées et 30% se sont retirés en raison de problèmes cutanés ou gastro-intestinauxNCT01171859 Phase II, ouverte, non randomisée, prospective 45
35 avec atteinte cardiaque ; 25 ATTRm ; 5 ATTRm avec TxH ; 13 ATTRwt ; et 2 transplantés hépatiques dominoDoxycycline 100 mg toutes les 12 h + TUDCA 250 mg toutes les 8 h pendant 12 mo
Phase de suivi subséquente sans traitement pendant 6 moRéponse au médicament définie comme une Réponse cardiaque évaluée chez 25 patients
68% avaient une réponse cardiaque
Augmentation du NT-.proBNP et détérioration de la souche pendant le suivi sans traitement
Nombre élevé d’abandons en raison d’effets indésirables
14 abandons en phase de traitement et 5 abandons en phase sans traitementEffet de la doxycycline + UDCA sur l’ATTR Phase II, ouverte, non randomisée, prospective 28
ATTR avec atteinte cardiaque (27 ATTRm et 1 ATTRwt)Doxycycline 200 mg/j pendant 4 wk, puis suspension 2 wk, puis UDCA 750 mg/j pendant 12 mo
Phase de suivi subséquente sans traitement pendant 6 moChangements dans le NT-proBNP et le score de Kumamoto Seulement 14% ont terminé l’étude et 36% ont terminé 12 mo
Aucun changement dans le NT-proBNP à 6 mo et aggravation à 12 mo
HVG stable
Augmentation du score de Kumamoto à 12 moEGCG NCT01171859 Phase II, ouverte, non randomisée, prospective 25
ATTRwt600 mg, EGCG pendant 12 mo Changements dans ECHO et IRM cardiaque (n = 14) Diminution de la masse LV 6% par IRM cardiaque (P = 0.03)
LVEF, épaisseur myocardique et MAPSE par ECHO inchangéesAntiSAP + CPHPC NCT03044353 Phase II, ouverte, randomisée 40
Cohorte 1 : amylose ATTR cardiaque
Cohorte 2 : amyloïdose primaire après 6 mo de chimiothérapieAnti-SAP + CPHPC mensuellement pendant 6 mo Réduction de la charge amyloïde par IRM cardiaque et ECHO Début en 2017 AntiSAP + CPHPC, antisérum composant P amyloïde + acide (R)-1–6-oxo-hexanoyl]pyrrolidine-2-carboxylique ; ATTRm, amyloïdose à transthyrétine mutante ; ATTRwt, amyloïdose à transthyrétine de type sauvage ; BNP, peptide natriurétique cérébral ; ECHO, échocardiogramme ; EGCG, épigallocatéchine-3 gallate ; IV, intraveineux ; LV, ventricule gauche ; FEVG, fraction d’éjection du ventricule gauche ; HVG, hypertrophie ventriculaire gauche ; MAPSE, excursion systolique du plan annulaire mitral ; mNIS, Modified Neuropathy Impairment Score ; IRM, imagerie par résonance magnétique ; NIS, Neuropathy Impairment Score ; NIS-LL, Neuropathy Impairment Score of the Lower Limbs ; NT-proBNP, amino-terminal pro-brain natriuretic peptide ; SC, sous-cutané ; TTR, transthyrétine ; TUDCA, acide tauroursodéoxycholique ; TxC, transplantation cardiaque ; TxH, transplantation hépatique ; UDCA, acide ursodéoxycholique.
Les AO sont de courts brins d’oligonucléotides qui se lient spécifiquement à l’ARN, empêchant la traduction et la synthèse des protéines cibles4. ISIS-TTRRX est un AO sous-cutané, avec des réductions dose-dépendantes démontrées des valeurs de TTR de 75% à 90% chez des volontaires sains.L’essai de phase III chez des patients atteints d’ATTRm et de phénotype neurologique s’est terminé en mars 2017 et ses résultats sont attendus pour la fin de l’année 2017. Cependant, la Food and Drug Administration américaine a reporté le lancement d’un essai de phase III chez les patients atteints d’ATTRwt et d’ATTRm avec cardiopathie en raison de cas de thrombocytopénie sévère dans l’étude neurologique (tableau 2). Comme 50 % des participants à l’étude neurologique présentaient une maladie cardiaque, les résultats de cette sous-étude cardiaque détermineront si l’essai de phase III est repris. D’autre part, il existe des données préliminaires d’un essai ouvert de phase II. Dans cette étude, 22 patients atteints d’ATTRwt et d’ATTRm avec une maladie cardiaque ont reçu une injection hebdomadaire d’ISIS-TTRRX. Selon le rapport, le profil de sécurité du médicament est très favorable et les données intermédiaires sur la progression de la maladie cardiaque par CMR, NT-proBNP et tests de 6 minutes sont positives.41
Stabilisation de la transthyrétine
La dissociation du tétramère TTR en sous-unités est une étape cruciale dans la formation des fibrilles ATTR. Le diflunisal et le tafamidis sont 2 stabilisateurs de la TTR ayant une efficacité démontrée dans la polyneuropathie ATTRm.
- –
Le tafamidis est une petite molécule administrée par voie orale qui se lie à la TTR au niveau des sites de liaison T4 en stabilisant la protéine et en empêchant sa dissociation. Suite à la publication des résultats d’un essai randomisé en double aveugle chez 125 patients atteints d’ATTRm et de la mutation Val30Met aux premiers stades de la maladie neurologique,42 l’Agence européenne des médicaments a approuvé son utilisation en 2011 en tant que médicament orphelin pour retarder la progression neurologique. Des données récentes démontrent l’efficacité du médicament pour atteindre une stabilité neurologique chez au moins 60 % des participants après plus de 4 ans de suivi. À ce jour, son utilisation est limitée à l’ATTR et aux maladies cardiologiques. Une étude de phase II chez 21 patients atteints d’ATTRm et de différentes mutations a montré que le NT-proBNP et les paramètres échocardiographiques restaient stables à 12 mois.43 Les données d’une étude de cohorte de 5 ans ont confirmé que le médicament était bien toléré à une dose de 20 mg, bien que peu de patients atteints d’ATTRwt soient restés stables à 3,5 ans.44 L’essai ATTR-ACT est un essai de phase III de 30 mois évaluant l’efficacité, la sécurité et la tolérance de doses de 20 mg et 80 mg de tafamidis par rapport à un placebo chez 440 patients atteints d’ATTRm, d’ATTRwt et d’HF. Le critère d’évaluation principal comprend la mortalité et l’admission à l’hôpital. Ses résultats sont attendus en 2018.3,27
- –
Diflunisal est un agent anti-inflammatoire non stéroïdien qui stabilise les molécules de TTR in vitro. Il n’est pas disponible en Espagne, mais peut faire l’objet d’une demande médicale à l’étranger pour un usage compassionnel. Une étude de phase III sur l’ATTRm chez des patients présentant une atteinte neurologique prédominante, dont plus de la moitié avaient une maladie cardiaque, n’a trouvé aucune différence significative dans les paramètres échocardiographiques au cours de la période d’étude (Tableau 2).45 Son potentiel d’effets indésirables gastro-intestinaux, d’insuffisance rénale, de rétention d’eau et d’hypertension le rend inadapté aux patients ayant une maladie cardiaque. Les données sur le diflunisal chez les patients atteints d’ATTR sont très limitées. Il existe une étude, mais elle est limitée par sa conception monocentrique non randomisée avec peu de suivi et peu de patients (n = 13). Il n’y a pas eu d’admission pour une HF décompensée, mais il y a eu une aggravation significative de la fonction rénale.46
- –
Plus récemment, un groupe espagnol a démontré que la tolcapone (un inhibiteur oral de la catéchol-O-méthyltransférase utilisé dans le traitement de la maladie de Parkinson) a la capacité de se lier in vitro à la TTR des patients atteints d’ATTRwt et Val122Ile avec une affinité plus élevée que les autres stabilisateurs.47
Elimination des dépôts amyloïdes
Les dépôts amyloïdes sont très stables et il semble que l’organisme humain ait peu de capacité à les éliminer. Cependant, les traitements qui empêchent la production de nouveaux amyloïdes, comme la chimiothérapie dans la SLA, peuvent éliminer progressivement les dépôts à des taux différents selon les organes. La clairance cardiaque est particulièrement faible et jusqu’à présent, les preuves de régression sont rares. Plusieurs molécules sont actuellement à l’étude pour accélérer la clairance cardiaque amyloïde dans l’ATTR :
- –
La doxycycline (un antibiotique couramment utilisé) perturbe la formation des fibrilles amyloïdes. L’effet synergique de l’association de la doxycycline et de l’acide biliaire tauroursodésoxycholique (TUDCA), utilisé dans le traitement des maladies du foie, a été démontré pour éliminer les dépôts de TTR dans des modèles animaux. Une étude de phase II portant sur 20 patients n’a montré aucune progression cardiaque ou neurologique après un an de traitement par doxycycline/TUDCA, avec un profil de sécurité et de tolérance acceptable.4 D’autres études de phase II ont tenté de confirmer ces résultats en utilisant la combinaison doxycycline/TUDCA, doxycycline/acide ursodésoxycholique ou doxycycline seule.48-50 Les résultats préliminaires de l’une de ces études suggèrent un effet protecteur, avec une moindre aggravation de la fonction cardiaque due à la tension dans le groupe traité. Une autre de ces études a obtenu des résultats similaires chez 40 patients atteints d’ATTR : les paramètres NT-proBNP, classe fonctionnelle, FEVG et épaisseur myocardique, entre autres, sont restés stables à 12 mois (tableau 2). Néanmoins, toutes ces études présentaient un taux d’abandon élevé (35%-44%), principalement en raison d’effets indésirables, en particulier une hypersensibilité au soleil et des plaintes gastro-intestinales (jusqu’à 30%).48-50
- –
L’EGCG (épigallocatéchine-3 gallate) est la catéchine la plus abondante dans le thé vert, et il a été démontré in vitro et dans un modèle murin qu’il inhibe la formation d’amyloïde et élimine les dépôts existants.4 L’IRMC a montré que l’administration quotidienne de 600 mg d’EGCG était associée à une stabilisation de la masse ventriculaire gauche dans un groupe de 25 patients (tableau 2).51
- –
Le PRX004 est un anticorps monoclonal qui agit en se liant à des épitopes spécifiques des monomères et à la TTR mal repliée. Il suscite ainsi l’élimination des dépôts en activant la phagocytose.52 La base de son mécanisme d’action est similaire à celle d’un anticorps utilisé dans la LA. Les études de phase II sur cet anticorps donnent des résultats prometteurs. Un essai de phase I-II de ce nouvel anticorps devrait débuter en 2017.
- –
Qu’importe le type de protéine précurseur amyloïde, tous les dépôts contiennent le composant P amyloïde sérique (SAP). En utilisant cette molécule comme cible, il a été démontré que les anticorps anti-SAP déclenchent une réaction médiée par les macrophages et dépendante du complément qui entraîne une élimination majeure des dépôts amyloïdes viscéraux dans un modèle murin. Le composé bis-D-proline CPHPC peut neutraliser le SAP plasmatique, et la co-administration avec des IgG anti-SAP permet à l’anticorps d’atteindre les dépôts contenant du SAP dans les tissus.53 Une étude de phase I publiée en 2015 a démontré l’élimination des dépôts hépatiques chez 15 patients atteints d’amylose systémique sans atteinte cardiaque, avec peu d’effets indésirables.53 Une étude de phase II portant sur des patients atteints d’amylose cardiaque ATTR et d’AL devrait débuter en 2017 (tableau 2).
CONCLUSIONS
L’amylose cardiaque à transthyrétine est diagnostiquée avec une fréquence croissante. La scintigraphie au 99mTc-DPD et l’IRMC sont des exemples de techniques qui peuvent être utilisées pour l’identification simple et précoce des patients atteints d’ATTR.
Plusieurs médicaments spécifiques de l’ATTR sont actuellement en phase finale de recherche. Par conséquent, nous pensons que l’amylose cardiaque ATTR sera bientôt considérée comme une entité traitable plutôt que comme une maladie mortelle.
FUNDING
Ce travail a été réalisé avec l’aide partielle de l’Institut de santé Carlos III et de la Société espagnole de cardiologie (bourse de recherche 2016 à E. González-López). L’assistance de l’Institut de santé Carlos III est financée par le Fonds européen de développement régional « Une autre façon de faire l’Europe ».
CONFLITS D’INTÉRÊTS
E. González-López a participé en tant que conférencier à des activités organisées par Pfizer. P. Garcia-Pavia a reçu des paiements en tant que conférencier dans des activités organisées par Pfizer et en tant que consultant pour Alnylam, Prothena et Pfizer. E. González-López, A. López-Sainz, et P. Garcia-Pavia déclarent que Pfizer a financé des projets de recherche de leur institution.