INTRODUCTION
Amyloidose ist eine Ablagerungskrankheit, die durch eine extrazelluläre Ansammlung von Fibrillen verursacht wird, deren Ursprung aus Proteinen mit einer instabilen Struktur besteht, die sich falten, aggregieren und ablagern.1 Solche Ablagerungen können die Gewebestruktur verändern und die Funktion verschiedener Organe und Systeme beeinträchtigen.2
Amyloidfibrillen sind unlöslich und proteolyseresistent und werden typischerweise mit Kongorot angefärbt und zeigen unter polarisiertem Licht eine intensive gelb-grüne Doppelbrechung.3 Mehr als 30 Proteine können Amyloidablagerungen verursachen, aber nur 5 verursachen signifikante Ablagerungen im Herzgewebe1:
- –
Leichtketten, die primäre Amyloidose (AL) verursachen.
- –
Transthyretin (TTR), das TTR-Amyloidose (ATTR) verursacht.
- –
Apolipoprotein A.
- –
Fibrinogen.
- –
Serum-Amyloid-Protein A, das eine sekundäre Amyloidose hervorruft.
Primäre Amyloidose und ATTR sind die häufigsten Formen der kardialen Amyloidose, wobei die AL-Form historisch gesehen in den Industrieländern häufiger vorkommt.3
Die meisten Informationen über die kardiale Amyloidose beziehen sich auf die AL. Doch obwohl die Zahl der Patienten mit AL stabil geblieben ist, hat die Zahl der ATTR-Diagnosen in letzter Zeit zugenommen, und man geht inzwischen davon aus, dass ATTR viel häufiger vorkommt als AL.2
Transthyretin-Amyloidose ist sehr häufig Gegenstand von Fehldiagnosen oder erheblichen Verzögerungen bis zur korrekten Diagnose. Gründe dafür sind die Heterogenität der Formen, die Notwendigkeit einer histologischen Bestätigung, der Mangel an Spezialgeräten und die irrige Meinung einiger Kardiologen, dass es sich um eine seltene Krankheit ohne Behandlungsmöglichkeiten handelt.2,3
Diese Aspekte ändern sich jedoch. Die Diagnose hat Auswirkungen auf die Behandlung der Patienten. Es wurden spezifische Therapien entwickelt, die die Ablagerung verzögern oder stabilisieren können und die in den frühen Stadien wirksamer sind. Eine frühzeitige Diagnose ist daher von entscheidender Bedeutung. In dieser Übersicht werden die jüngsten Fortschritte bei der Diagnose und Behandlung von ATTR beschrieben, die Patienten mit dieser Erkrankung Hoffnung geben.
TRANSTHYRETIN CARDIAC AMYLOIDOSIS
Transthyretin ist ein tetrameres Plasmaprotein, das für den Transport von Thyroxin und retinolgebundenem Protein verantwortlich ist. Es wird primär in der Leber und sekundär im Plexus choroideus und im Pigmentepithel der Netzhaut synthetisiert.4
Transthyretin neigt dazu, in Dimere und Monomere zu dissoziieren, die sich zu Fibrillen zusammenlagern und ablagern. Punktmutationen oder der Einfluss des Alters können diese Tendenz verstärken, was zu den beiden klinischen Formen von ATTR führt: mutiert (ATTRm) und Wildtyp (ATTRwt).
MUTANT TRANSTHYRETIN AMYLOIDOSIS
Derzeit sind mehr als 120 Mutationen bekannt, die ATTRm verursachen. Diese Mutationen zeigen ein autosomal-dominantes Vererbungsmuster mit unterschiedlicher Penetranz.4 Aufgrund der großen geografischen Vielfalt ist es schwierig, die Prävalenz von ATTR zu bestimmen, aber sie gilt als seltene Krankheit mit einer Prävalenz von weniger als 1/100 000 Einwohner2 (Tabelle 1).
Hauptmerkmale der klinischen und diagnostischen Merkmale von Mutanten- und Wild-.Typ Transthyretin Cardiac Amyloidosis
| ATTRwt | ATTRm | |
|---|---|---|
| Prävalenz | Unbekannt. Anscheinend sehr häufig | |
| Genetische Untersuchung | Abwesenheit von Mutationen in TTR | Mutation in TTR |
| Typisches Alter bei der Präsentation | > 60 Jahre | Variabel je nach ursächlicher Mutation |
| Geschlecht | Männliches Übergewicht. 80% der Patienten | Männlich vorherrschend, mit aggressiverem Phänotyp |
| Extrakardiale Manifestationen | – Karpaltunnelsyndrom (33%-49%) – Lumbale Spinalkanalstenose – Traumatische Bizepssehnenruptur (32%) |
– Aufsteigende bilaterale sensorisch-motorische Polyneuropathie – Dysautonomie: Orthostatische Hypotonie, Diarrhöe-Verstopfung, erektile Dysfunktion – Augenbeteiligung: Glaukom, intravitreale Ablagerungen, |
| Kardiale Beeinträchtigung | Konstant | Variabel je nach ursächlicher Mutation |
| Herzleistung | – Herzinsuffizienz (53%-86%) – Erregungsleitungsstörungen – Vorhofflimmern (43%-67%) – Degenerative AoS |
– Überleitungsstörungen – Herzinsuffizienz – Seltenes Vorhofflimmern (10%) |
| Diagnostische Verfahren | ||
| EKG | – Pseudoinfarktmuster (63%-66%) – Niedrige Spannung (22%-33%) – Sokolow LVH (6%-13%) |
– Pseudoinfarktmuster (18%-69%) – Niedrige Spannung (2%-25%) – Sokolow LVH (3%-8%) |
| ECHO | – Mäßig-schwere Hypertrophie – Mild-mäßig depressive LVEF (30%) |
– Mäßige Hypertrophie – LVEF, typischerweise erhalten |
| Herz-MRT | – Spätes Enhancement – Erhöhtes natives T1 und EV |
|
| 99mTc DPD-Szintigraphie | – Grad 2-3 | – Grad 0: asymptomatische Träger – Grad 1: anfängliche kardiale Beteiligung – Grad 2-3: deutliche Herzbeteiligung |
AF, Vorhofflimmern; AoS, Aortenstenose; ATTRm, mutierte Transthyretin-Amyloidose; ATTRwt, Wildtyp-Transthyretin-Amyloidose; EKG, Elektrokardiogramm; ECO, Echokardiogramm; EV, Extrazellularvolumen; LVEF, linksventrikuläre Auswurffraktion; LVH, linksventrikuläre Hypertrophie; TTR, Transthyretin.
Die ersten TTR-Mutationen wurden als familiäre Amyloid-Polyneuropathie (oder Andrade-Krankheit) gemeldet, und folglich wurde ATTRm bis vor kurzem als neurologische Erkrankung betrachtet. Neuere Erkenntnisse zeigen jedoch, dass in mehr als der Hälfte der Fälle das Herz betroffen ist.3
Es besteht eine starke Genotyp-Phänotyp-Korrelation, wobei Mutationen mit rein neurologischen Erkrankungen oder rein kardialen Erkrankungen assoziiert sind.3 Die Einteilung von ATTRm in eine kardiale oder neurologische Erkrankung könnte jedoch eine zu starke Vereinfachung sein, da es zwischen den beiden klinischen Formen des Krankheitsspektrums erhebliche Überschneidungen gibt.
Die Val30Met-Mutation (jetzt als Val50Met bekannt, nachdem dem traditionellen Mutationsnamen bei ATTRm 20 Positionen hinzugefügt wurden) ist die häufigste Mutation weltweit und kommt in Portugal, Japan und Schweden vor. Die geschätzte Inzidenz in Portugal beträgt 1 pro 538 Einwohner.2 Mallorca (Spanien) und Valverde del Camino (Huelva, Spanien) gelten ebenfalls als Gebiete, in denen ATTRm endemisch ist. Die geschätzte Prävalenz auf Mallorca bei symptomatischen Patienten liegt bei 3/100 000 Einwohnern.5
Die Val30Met-Mutation verursacht eine vorwiegend neurologische Erkrankung mit symmetrischer sensomotorischer Polyneuropathie, die in den unteren Gliedmaßen beginnt und einem aufsteigenden Muster folgt. Sie kann mit Dysautonomie mit orthostatischer Hypotonie, erektiler Dysfunktion, Harninkontinenz und gastrointestinalen Symptomen einhergehen. Sie beginnt typischerweise am Ende des zweiten oder dritten Lebensjahrzehnts, und bis zu 43 % der Patienten haben eine kardiale Beteiligung, die eine häufige Todesursache ist4 (Tabelle 1).
Von besonderer Bedeutung ist die Val122Ile-Mutation (p. Val142Ile), die bei 3 % bis 4 % der schwarzen Bevölkerung Nordamerikas vorkommt.3 Obwohl die Penetranz unvollständig ist,3 wurde diese Mutation mit einem um 47 % erhöhten Risiko für die Entwicklung einer Herzinsuffizienz (HF) in Verbindung gebracht.6 Eine kürzlich durchgeführte Studie zeigte, dass die Val122Ile-Amyloidose die vierthäufigste Ursache für HF in der britischen afro-karibischen Bevölkerung war.7 Obwohl bis zu 30 % der Patienten mit dieser Mutation Merkmale einer leichten Neuropathie aufweisen können,6 ist der klinische Phänotyp in der Regel ähnlich dem von ATTRwt.4 Val122Ile sollte nicht als eine Mutation angesehen werden, die ausschließlich in der schwarzen Bevölkerung vorkommt, da sie auch in der weißen Bevölkerung vorhanden sein kann. Wir haben diese Mutation zum Beispiel in 4 weißen spanischen Familien ohne schwarze Vorfahren identifiziert.
Wildtyp-Transthyretin-Amyloidose
Die Wildtyp-Transthyretin-Amyloidose wurde erstmals 1876 beschrieben. Früher wurde sie als senile Amyloidose bezeichnet, aber da sie bei Patienten im Alter von 40 bis 60 Jahren diagnostiziert wird, ist diese Bezeichnung inzwischen überholt. Interessanterweise wurde der früheste bekannte Fall dieser Mutation bei einem 47-jährigen amerikanischen Patienten gefunden.8
Die genaue Prävalenz von ATTRwt ist nach wie vor unbekannt. Studien deuten jedoch darauf hin, dass sie unterdiagnostiziert ist und möglicherweise die häufigste Form der kardialen Amyloidose darstellt.2,3 Die folgenden Ergebnisse unterstützen diese Hypothese:
- –
Bei Patienten im Alter von über 80 Jahren liegt die Prävalenz von TTR-Ablagerungen bei der Autopsie bei 25%.3
- –
Bei Patienten mit HF mit erhaltener Ejektionsfraktion (HFpEF) liegt die Prävalenz mittelschwerer TTR-Ablagerungen bei der Autopsie bei 5 %.9
- –
Bei Patienten im Alter von über 60 Jahren, die wegen HFpEF und einer linksventrikulären Hypertrophie (LVH) ≥ 12 mm aufgenommen wurden, fand unsere Gruppe kürzlich eine Prävalenz von 13 %.10
Im Gegensatz zu ATTRm ist ATTRwt eine sporadische Erkrankung, die typischerweise nach dem Alter von 70 Jahren beginnt.4 Sie tritt hauptsächlich bei Männern auf, und in veröffentlichten Serien wurde von Raten zwischen 89 % und 98 % berichtet.11,12 In einer kürzlich durchgeführten Serie von Patienten, bei denen ATTRwt in zwei Krankenhäusern (Madrid, Spanien und Bologna, Italien) diagnostiziert wurde, stellte unsere Gruppe jedoch fest, dass 20 % Frauen waren. Darüber hinaus haben auch andere Autopsiestudien ergeben, dass ATTRwt bei Frauen weiter verbreitet sein könnte als bisher berichtet. Daher sollte das weibliche Geschlecht den klinischen Verdacht auf ATTRwt nicht mindern (Tabelle 1).13
Autopsieergebnisse zeigen, dass die TTR-Ablagerungen bei ATTRwt in verschiedenen Organen verteilt sind. Aufgrund des kardialen Tropismus von TTR ist die Ablagerung im Herzen jedoch viel größer, und die kardiale Beteiligung ist die wichtigste klinische Manifestation.4 Die Patienten können Symptome einer extrakardialen TTR-Ablagerung aufweisen, wie z. B. Lumbalkanalstenose, atraumatische Ruptur der Bizepssehne oder „Popeye-Zeichen“ und Karpaltunnelsyndrom (CTS)3 (Abbildung 1). All diese Merkmale können dazu beitragen, die Diagnose zu stellen und schnell zu ermitteln. Das CTS kann auch bei anderen Amyloidose-Subtypen auftreten, ist aber bei ATTRwt häufiger. Die Ablagerung kann den kardialen Manifestationen um mehrere Jahre vorausgehen.6 Es kann als Indikation bei älteren Patienten mit LVH verwendet werden, insbesondere wenn sie ein beidseitiges CTS haben, das nicht mit spezifischen beruflichen Aktivitäten in Verbindung steht und in die Funktionsklasse ≥ II der New York Heart Association fällt (unveröffentlichte Daten).
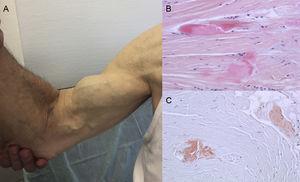
Anzeichen und Symptome der Transthyretin-Amyloidose. A: nicht-traumatische Ruptur der rechten Bizepssehne („Popeye-Zeichen“). B und C: Färbung mit Hämatoxylin-Eosin (B) und Kongorot (C), beide ×200, einer Probe des Karpalbandes, die dichte Kollagenbündel mit nichtzellulärem Material zeigt. Mit freundlicher Genehmigung von Dr. Clara Salas Antón.
DIAGNOSE DER TRANSTHYRETIN-AMYLOIDOSISKlinische Darstellung
Amyloid kann jede kardiale Struktur infiltrieren.1 Typischerweise erhöht sich durch die Ablagerung die Wanddicke des Ventrikels, was zu einer allmählichen Abnahme der Dehnbarkeit und damit zu einer schweren diastolischen Dysfunktion führt. ATTR wird daher traditionell als Ursache einer restriktiven Kardiomyopathie angesehen.
Das klinische Spektrum von ATTR ist jedoch viel breiter und heterogener. Das häufigste Symptom von ATTR ist HF. Wie bereits erwähnt, deutet eine 2015 von unserer Gruppe veröffentlichte Studie darauf hin, dass ein auf 99mTc-3,3-Diphosphono-1,2-Propanodicarbonsäure (99mTc-DPD)-Szintigraphie basierendes Protokoll für die Diagnose von ATTRwt bei einem signifikanten Anteil (13 %) von Patienten, die älter als 60 Jahre sind und wegen HFpEF eingewiesen wurden, nützlich sein kann.10 Aufgrund dieses Ergebnisses wurde die 99mTc-DPD-Szintigraphie in die europäischen Leitlinien für HF von 2016 als nützliches Instrument zur Identifizierung von Patienten mit ATTR aufgenommen.14 ATTR sollte jedoch nicht ausschließlich bei Patienten mit HFpEF vermutet werden, da sich mit fortschreitender Amyloidablagerung die kontraktile Funktion verschlechtert und ATTR folglich mit unterschiedlichen Graden systolischer Dysfunktion assoziiert sein kann.
Transthyretin-Amyloidose ist eine Phänokopie der hypertrophen Kardiomyopathie (HCM) und kann mit dieser verwechselt werden. In einer kürzlich durchgeführten multizentrischen französischen Studie wurde berichtet, dass 5 % der Patienten mit HCM ATTRm haben.15 Unsere Ergebnisse stimmen jedoch nicht mit dieser hohen Rate überein, was mit der großen schwarzen Bevölkerung in Frankreich zusammenhängen könnte.
Kardiale Erregungsleitungsanomalien können die erste Manifestation von ATTR sein. Die Amyloidinfiltration des Sinus und der atrioventrikulären Knoten1 kann auf die Notwendigkeit einer Schrittmacherimplantation hinweisen (Tabelle 1). In der bereits erwähnten Studie, die in Spanien und Italien durchgeführt wurde, wurde festgestellt, dass Erregungsleitungsstörungen bei 7 % der Patienten mit ATTRwt die erste Manifestation dieser Krankheit waren.13
Auch Herzrhythmusstörungen sind bei Patienten mit ATTRwt sehr häufig13 (Abbildung 2A), und die erste Manifestation der Krankheit kann ein Schlaganfall sein.4 Die Mayo Clinic-Gruppe hat kürzlich vorgeschlagen, dass ATTRwt bei der Diagnose von nichtvalvulärem Vorhofflimmern (AF) bei älteren Patienten ausgeschlossen werden sollte.8 Im Gegensatz dazu ist AF bei Patienten mit ATTRm viel seltener (Tabelle 1).
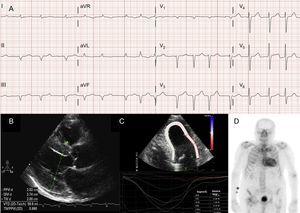
Diagnostische Verfahren bei Transthyretin-Herzamyloidose (ATTR). A: Elektrokardiogramm eines Patienten mit Wildtyp-Transthyretin-Amyloidose (ATTRwt), das Vorhofflimmern und Pseudoinfarktmuster in den unteren Ableitungen zeigt. B: Echokardiogramm eines Patienten mit mutierter Transthyretin-Amyloidose mit Val30Met-Mutation, mit ausgeprägter konzentrischer linksventrikulärer Hypertrophie und leichtem Perikarderguss. C: longitudinale regionale Belastung eines Patienten mit ATTRwt, mit erhaltenen Werten im apikalen Segment und erniedrigten Werten im basalen und mittleren ventrikulären Segment. D: 99mTc-DPD (99mTc-3,3-Diphosphono-1,2-Propanodicarbonsäure)-Scan eines Patienten mit ATTRwt, der eine biventrikuläre Aufnahme zeigt, die der Knochenaufnahme überlegen ist und dem Perugini-Grad 3 entspricht.
Abschließend stellen wir fest, dass ATTR und degenerative Aortenstenose bei ein und demselben Patienten nebeneinander bestehen können. Im Jahr 2016 machten mehrere Studien auf diese Möglichkeit aufmerksam, und in einer prospektiven Studie wurde berichtet, dass ATTRwt bei Patienten über 65 Jahren, bei denen ein Aortenklappenersatz vorgenommen wurde, eine Prävalenz von 6 % aufweist.16 Diese Studie deutet darauf hin, dass Patienten mit beiden Entitäten eine wesentlich schlechtere postoperative Prognose haben als Patienten ohne ATTRwt (Sterblichkeit 50 % gegenüber 6,9 % nach einer medianen Nachbeobachtungszeit von 2,3 Jahren).16 In einer weiteren aktuellen Studie mit 99mTc-DPD-Szintigraphie bei 43 Patienten mit Aortenstenose mit niedrigem Fluss und niedrigem Gradienten wurden 5 Patienten mit ATTRwt identifiziert (Prävalenz 12 %). 17 Patienten mit schwerer Aortenstenose und ATTRwt weisen das gleiche demografische Profil auf, und die geeignete Behandlung für Patienten mit beiden Erkrankungen muss noch ermittelt werden.
Nutzen diagnostischer Techniken
Die Diagnose von ATTR ist eine Herausforderung in der täglichen klinischen Praxis. Obwohl Elektrokardiographie und Echokardiographie bei der Diagnose eine Rolle spielen, haben neue nicht-invasive Techniken eine Schlüsselrolle bei der Beurteilung von Patienten mit ATTR erlangt.
Elektrokardiogramm
Der Zusammenhang zwischen Niederspannung und kardialer Amyloidose gilt seit langem als unbestreitbar.3 Die in der klinischen Praxis am häufigsten verwendeten Kriterien sind die QRS-Amplitude 1 Obwohl niedrige elektrokardiografische Spannungen bei LVH einen Verdacht begründen sollten, lag die Prävalenz in einer zeitgenössischen Serie von ATTR-Patienten bei nur 20 bis 25 %.3,4,13 Die Prävalenz variiert auch je nach den verwendeten Kriterien. So kann die Anwendung des Sokolow-Kriteriums (S-Welle in Ableitung V1 + R-Welle in Ableitung V5 oder V6
1,5 mV) die berechnete Prävalenz auf 46 % bis 58 % erhöhen.13 Das Verhältnis der linksventrikulären Wanddicke zur gesamten QRS-Spannung wurde empfohlen, um Unterschiede zwischen den Ergebnissen der beiden Techniken besser beurteilen zu können.2,3 Allerdings können bis zu 20 % der Patienten mit ATTR die elektrokardiographischen Kriterien für LVH erfüllen.2,3
In den meisten Serien von Patienten mit kardialer Amyloidose ist das Pseudoinfarktmuster der häufigste elektrokardiographische Befund2,3,13 (Abbildung 2A). Aufgrund einer möglichen Beteiligung des Reizleitungssystems sind auch komplette oder inkomplette Schenkelblöcke häufig.3
Echokardiographie
Obwohl die Echokardiographie der Eckpfeiler der Erstdiagnose von ATTR ist, sind die Befunde nicht spezifisch.3 Die Transthyretin-Amyloidose ist typischerweise mit einem normalen oder kleinen linken Ventrikel mit konzentrischer Hypertrophie verbunden.3 Auf dem 10. internationalen Symposium über Amyloid und Amyloidose im Jahr 2004 wurde als echokardiographisches Kriterium für eine Herzerkrankung aufgrund von AL bei Fehlen anderer Ursachen für LVH das Vorhandensein von LVH mit einem Grenzwert von 12 mm für die interventrikuläre Septumwanddicke festgelegt.4 Dieses Kriterium wurde später auf andere Formen der Amyloidose extrapoliert (Abbildung 2B), was zu einer hohen Spezifität, aber einer geringen Sensitivität führte.
Obwohl klassischerweise eine konzentrische LVH beschrieben wurde, deuten aktuelle Serien darauf hin, dass etwa 20 % eine asymmetrische LVH aufweisen.13
Trotz der klassischen Assoziation zwischen einer normalen oder leicht verringerten linksventrikulären Auswurffraktion (LVEF) und kardialer Amyloidose,2 ist der LVEF-Bereich sehr variabel.8 In einer kürzlich an der Mayo Clinic durchgeführten Studie lag die LVEF bei 8, während in unserer Serie die LVEF bei 13 lag. Darüber hinaus ist die Verwendung der LVEF bei der Beurteilung der systolischen Funktion bei kardialer Amyloidose begrenzt, da leicht erniedrigte Werte bereits auf eine relevante kardiale Erkrankung hinweisen. Diese Einschränkung kann durch die Verwendung von Gewebe-Doppler-Geschwindigkeiten, Dehnungsbildern und der myokardialen Kontraktionsfraktion überwunden werden, die als geeignetere Indizes zur Beurteilung der Herzfunktion vorgeschlagen wurden.2
Weitere klassische echokardiographische Zeichen sind rechtsventrikuläre Hypertrophie, biatriale Dilatation, leichter Perikarderguss, Verdickung der Atrioventrikularklappe, Wandverdickung der Vorhofscheidewand und körniges, funkelndes Aussehen des Myokards.3,6 Da jedoch einige dieser Merkmale in einer hochselektierten Serie von Patienten in fortgeschrittenen Krankheitsstadien beobachtet wurden, müssen nicht alle von ihnen vorhanden sein, um einen Verdacht zu begründen.1
Die regionale Dehnungsbildgebung ist eine sehr nützliche Technik für die Frühdiagnose von Patienten mit ATTR. Bei Patienten mit ATTR ist die longitudinale Dehnung in den basalen und mittleren ventrikulären Segmenten vermindert, während sie in den apikalen Segmenten erhalten bleibt18 (Abbildung 2C). Dieses typische Muster kann bei der Differenzialdiagnose von ATTR gegenüber anderen Herzerkrankungen hilfreich sein.4
Biomarker
Es gibt weniger Daten über die Rolle des N-terminalen Prohormons des natriuretischen Propeptids des Gehirns (NT-proBNP) und Troponin bei ATTR als bei AL.4 Die NT-proBNP-Spiegel bei ATTR sind in der Regel niedriger als bei AL,4 was zwei unterschiedliche pathophysiologische Mechanismen widerspiegelt: direkte Leichtketten-Toxizität bei AL vs. induzierte Gewebeschäden durch Protofibrillen bei ATTR.
Kürzlich schlug die Gruppe der Mayo Clinic ein ähnliches Stratifizierungssystem vor, wie es für AL gilt. In einer Kohorte von 360 Patienten mit ATTRwt erwiesen sich beide Biomarker als Prädiktoren für die Mortalität. Patienten im Stadium III (NT-proBNP > 3000 pg/ml und Troponin T > 0,05 ng/ml) hatten ein medianes Überleben von 20 Monaten, während Patienten im Stadium I und II ein medianes Überleben von 66 Monaten bzw. 40 Monaten hatten (kein Biomarker bzw. nur ein Biomarker über den festgelegten Grenzwerten).
Kardiale Magnetresonanztomographie
Mit der kardialen Magnetresonanztomographie (CMRI) können strukturelle und funktionelle Informationen gewonnen und die Zusammensetzung des Herzmuskelgewebes charakterisiert werden.3 Die CMRI ist für die frühzeitige Erkennung von ATTR und für die Differenzialdiagnose zu anderen Herzerkrankungen von entscheidender Bedeutung.
Die Charakterisierung des Gewebes durch CMRI basiert auf den folgenden Merkmalen:
- –
Spätes Enhancement: Ein globales subendokardiales Muster ist praktisch pathognomonisch für eine kardiale Amyloidose, liegt aber nur bei etwa einem Viertel der Patienten vor. Andere Muster, wie z. B. transmurales (das häufigste) oder Patching, sind ebenfalls kompatibel (Abbildung 3). Trotz der hohen Sensitivität und Spezifität sollte berücksichtigt werden, dass möglicherweise keine späte Anreicherung vorhanden ist (15 % der Patienten) und unserer Erfahrung nach ein nicht zu vernachlässigender Prozentsatz an falsch-negativen Ergebnissen aus technischen Gründen auftritt.3 Das transmurale Anreicherungsmuster ist mit einer schlechteren Prognose verbunden und ein unabhängiger Prädiktor für die Mortalität.19
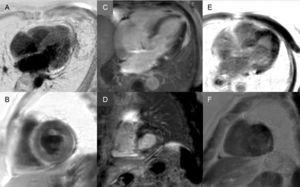 Abbildung 3.
Abbildung 3.Vielfalt der späten Enhancementmuster in der kardialen Magnetresonanztomographie bei Transthyretin-Amyloidose. A und B: Late-Enhancement-Sequenzen, 4-Kammer-Ebene bzw. kurze Achse auf mittlerer Höhe, eines Patienten mit mutierter Transthyretin-Amyloidose (ATTRm), die diffuse pathologische transmurale Gadoliniumablagerungen zeigen. C und D: späte Enhancement-Sequenzen, 4-Kammer- bzw. Kurzachsen-Basalebene, von Patienten mit ATTRm, die pathologische Gadoliniumablagerungen mit einem fleckigen Muster zeigen, mit einem unteren inferoseptalen und inferolateralen basalen Herdbereich. E und F, späte Enhancement-Sequenzen, 4-Kammer-Ebene bzw. kurze Achse auf der apikalen Ebene von Patienten mit ATTRm, die ausgedehnte pathologische transmurale Ablagerungen zeigen, außer in basalen und mittleren anterolateralen Segmenten. Mit freundlicher Genehmigung von Dr. Jesús González Mirelis.
(0.15MB). - –
Lange T1-Zeiten: Das T1-Mapping ist eine Technik, bei der ein quantitatives Herzmuskelsignal vor (natives T1) oder nach Kontrastmittelgabe gemessen wird. Native T1-Zeiten sind bei kardialer Amyloidose sehr lang.3 T1-Mapping erfordert keine Kontrastmittelgabe und kann daher bei Nierenversagen eingesetzt werden. Die T1-Zeiten können sogar abnormal sein, bevor eine LVH beobachtet wird.3 Die T1-Zeiten sind bei ATTR länger als bei HCM und Kontrollen (1097 ms ± 43 ms vs. 1026 ms ± 64 ms vs. 9,67 ms ± 34 ms; P
ms ± 68 ms; P = 0,01).20
Die Verabreichung von Kontrastmittel kann zur Berechnung des extrazellulären Volumens (ECV) und zur Bewertung der Zunahme des extrazellulären Raums verwendet werden. ECV-Werte bei kardialer Amyloidose sind höher als bei anderen Herzerkrankungen, außer in Myokardinfarktzonen.21 Im Jahr 2016 berichtete unsere Gruppe in Zusammenarbeit mit anderen nationalen Zentren, dass die ECV-Quantifizierung eine kardiale Beteiligung bei ATTRm identifizieren kann und zum ersten Mal mit dem Grad der neurologischen Beeinträchtigung korreliert, was die Verwendung dieser Technik bei der Frühdiagnose und Verfolgung von ATTRm unterstützt.22
Quantitative T1-Mapping- und ECV-Berechnungstechniken können bei ATTR für die Frühdiagnose, die klinische Verlaufskontrolle und die Beurteilung des Therapieansprechens sehr nützlich sein (Abbildung 4).
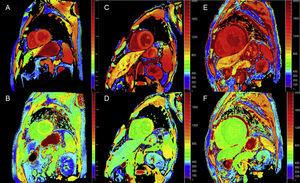
T1-Mapping, vor und nach Kontrastierung, mit modifizierter Look-Locker-Inversion-Recovery (MOLLI) in der kardialen 3T-Magnetresonanztomographie bei gesunden Kontrollpersonen, Patienten mit Transthyretin-Amyloidose und Patienten mit primärer Leichtketten-Amyloidose. A und B: Natives T1-Mapping bzw. extrazelluläres Volumen (EV) bei einer gesunden Kontrollperson mit normalen Werten (EV = 0,214). C und D: Natives T1-Mapping bzw. EV bei einem Patienten mit mutierter Transthyretin-Amyloidose mit neurologischen Schäden und beginnender kardialer Beteiligung, erhöhtem nativen T1 und leicht erhöhtem EV (0,361). E und F: Natives T1-Mapping bzw. EV bei einem Patienten mit kardialer Wildtyp-Transthyretin-Amyloidose, erhöhtem nativen T1 und sehr hohem EV (0,626), was auf eine massive Amyloid-Infiltration hinweist. Mit freundlicher Genehmigung von Dr. Jesús González Mirelis.
Herzszintigraphie
In den 1980er Jahren wurde die Beobachtung der kardialen Aufnahme verschiedener Knochendiphosphonat-Tracer histologisch mit dem Vorhandensein einer kardialen Amyloidose korreliert.23 Der Mechanismus der Aufnahme ist nicht gut charakterisiert, könnte aber mit dem Kalziumgehalt der Amyloidablagerungen zusammenhängen.
In einer frühen Studie der Bologna-Gruppe, in der 99mTc-DPD verwendet wurde, wurde bei 15 Patienten mit ATTR eine kardiale Aufnahme und bei 10 Patienten mit AL eine fehlende Aufnahme festgestellt, wobei ein auf der biventrikulären Aufnahme basierender Score verwendet wurde, der der Knochenaufnahme gleich oder überlegen war (Perugini-Score)24 (Abbildung 2D). Ähnliche Ergebnisse wurden anschließend von unserer Gruppe und anderen berichtet.25 Eine leichte (Score 1) und eine mäßige (Score 2) Anreicherung findet sich bei 30 % bzw. 10 % der Patienten mit AL.24
Aufgrund ihrer hohen Sensitivität und Spezifität ist diese Technik äußerst nützlich für die Diagnose von ATTR und kann eine kardiale Beteiligung auch dann zeigen, wenn Echokardiographie und MRT-Befunde noch normal sind. Nach einer Szintigraphie bei onkologischen oder rheumatologischen Indikationen sind zufällige ATTR-Befunde keine Seltenheit.26
Das Tc-DPD ist in den Vereinigten Staaten nicht erhältlich, aber es wurde über ähnliche Ergebnisse bei der Verwendung von 99mTc-PYP (Pyrophosphat)-Bildgebung berichtet.27
Weitere Radiotracer werden derzeit untersucht. So wurde beispielsweise 18F-Florbetapir, das bereits für die Darstellung von Beta-Amyloid im Gehirn zugelassen ist,4 bei Patienten mit AL und ATTR untersucht. Die Ergebnisse zeigen, dass 18F-Florbetapir myokardiale AL- und ATTR-Ablagerungen nachweisen kann.28 Obwohl die verfügbaren Daten in Fallstudien29 gewonnen wurden und die hohen Kosten dieses Radiotracers seine Verwendung einschränken, werden derzeit mehrere Studien zu den potenziellen Vorteilen seiner Verwendung gegenüber Tc-DPD als Screening-Technik für die beiden häufigsten Arten von Amyloidose durchgeführt.
Invasive Diagnose
Die endgültige Diagnose von ATTR basiert auf dem histologischen Nachweis von Amyloidfibrillen. Obwohl es auch extrakardiale Ablagerungen geben kann, variiert die Wahrscheinlichkeit des histologischen Nachweises von Amyloid je nach Organ.2 Es gibt nur wenige Studien über die Kosteneffizienz der extrakardialen Biopsie (z. B. Bauchfett, Zahnfleisch, Speicheldrüse, Magen-Darm) bei ATTR, die bei ATTRm größer ist als bei ATTRwt. Eine negative Biopsie eines klinisch nicht betroffenen Organs schließt jedoch die Diagnose ATTR nicht aus.4
Wie bei ATTRwt ist die Endomyokardbiopsie bei Patienten ohne extrakardiale Beteiligung oder mit alleiniger Herzerkrankung indiziert.3,4 Die Endomyokardbiopsie ist ein risikoarmes Verfahren (insbesondere in erfahrenen Zentren), und Entnahmefehler sind unwahrscheinlich.6
Nach der histologischen Bestätigung der Amyloidose, die manchmal die Interpretation durch geschultes Personal erfordert,6 ist die korrekte Klassifizierung des Subtyps von entscheidender Bedeutung.4 Derzeit hängt die Klassifizierung von einer Kombination aus Immunhistochemie, genetischer Analyse und Proteomik ab:
- –
Die Immunhistochemie basiert auf der Verwendung spezifischer Antikörper gegen bekannte Amyloidproteine. Obwohl die Ergebnisse dieser Technik in der Regel eindeutig sind, ist sie weniger empfindlich bei der Erkennung von Leichtketten.4
- –
Diese Einschränkung kann durch den Einsatz der Massenspektrometrie überwunden werden, die eindeutige Ergebnisse liefert und das Standardkriterium für die Bestätigung des Amyloid-Subtyps ist.2 Obwohl diese Technik nur in spezialisierten Zentren zur Verfügung steht, ist sie besonders nützlich bei nicht eindeutigen Fällen oder bei Fällen, in denen mehrere Antikörper in der Immunhistochemie positiv sind, was unserer Erfahrung nach in etwa 20 bis 30 % der Fälle der Fall ist. 4
- –
Da klinische oder histologische Verfahren ATTRm nicht von ATTRwt unterscheiden können, werden in allen ATTR-Fällen genetische Untersuchungen empfohlen. Die Feststellung einer ursächlichen Mutation kann für die genetische Beratung und Nachsorge von asymptomatischen Trägern von Bedeutung sein, 4,30 die von künftigen Therapien profitieren könnten, die den Ausbruch der Krankheit verzögern oder sogar verhindern.31
Nichtinvasive Diagnose
Bis vor kurzem galten histologische Untersuchungen als unverzichtbar für die Diagnose von ATTR.3 Um die Diagnose zu erleichtern, schlug eine internationale Multicenterstudie im Jahr 2016 einen neuen Algorithmus für die nichtinvasive Diagnose von ATTR vor.32
In der Studie wurden die Ergebnisse von 1217 Patienten analysiert. Das Vorhandensein klassischer Anzeichen einer kardialen Amyloidose unter Verwendung bildgebender Verfahren, eine Tc-DPD/PYP-Aufnahme von Grad 2 oder 3 in der Szintigrafie und das Fehlen eines monoklonalen Proteins hatten eine Spezifität und einen positiven Vorhersagewert für ATTR von 100 %32 (Abbildung 5).
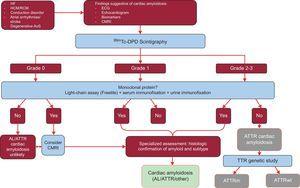
Diagnostischer Algorithmus für Patienten mit Verdacht auf kardiale Amyloidose. Einstufungssystem für die 99mTc-DPD-Szintigraphie: Grad 0, keine kardiale Aufnahme; Grad 1, geringfügig geringere Aufnahme als im Knochen; Grad 2, mäßige Aufnahme, die der Aufnahme im Knochen entspricht; Grad 3, starke Aufnahme, die der Aufnahme im Knochen überlegen ist. ACV, Schlaganfall; AL, primäre Leichtketten-Amyloidose; AoS, Aortenstenose; ATTR, Transthyretin-Amyloidose; ATTRm, mutierte Transthyretin-Amyloidose; ATTRwt, Wildtyp-Transthyretin-Amyloidose; CMRI, kardiale Magnetresonanztomographie; EKG, Elektrokardiogramm; HCM, hypertrophe Kardiomyopathie; HF, Herzinsuffizienz; RCM, restriktive Kardiomyopathie; TTR, Transthyretin.
Ein wesentliches Merkmal dieses Algorithmus ist das Fehlen eines monoklonalen Proteins, das beim Serumkettentest (Freelite, The Binding Site, UK) und bei der Immunofixationselektrophorese von Blut und Urin AL verursachen könnte. Das Vorhandensein eines monoklonalen Proteins ist eine Indikation für eine Endomyokardbiopsie zur Unterscheidung zwischen ATTR und AL.32 Bis zu 5 % der Bevölkerung im Alter von über 65 Jahren haben eine monoklonale Gammopathie von unbestimmter Signifikanz.2 Bei älteren Menschen sollte ein moderater Anstieg der zirkulierenden Leichtketten nicht direkt zur Diagnose einer AL führen. Es wurde berichtet, dass bis zu 10 % der älteren Patienten mit ATTRwt und monoklonaler Gammopathie unbestimmter Signifikanz in Referenzzentren zuvor eine Fehldiagnose von AL erhalten hatten.3,33 Eine korrekte Diagnose ist notwendig, um eine unangemessene Chemotherapie zu vermeiden. Interessanterweise hat unsere Klinik 2 Fälle von Patienten mit multiplem Myelom und gleichzeitiger ATTRwt in der Massenspektrometrie dokumentiert.
Behandlung von TRANSTHYRETIN CARDIAC AMYLOIDOSIS
Die Behandlung von Patienten mit ATTR hat zwei Ziele: medizinische Unterstützung und, wenn möglich, Stoppen oder Verzögern der Amyloidablagerung durch den Einsatz spezifischer Behandlungen.
Medizinische Behandlung
Die folgenden Abschnitte beschreiben die unterstützende kardiale Versorgung von Patienten mit ATTR.
Management der Herzinsuffizienz
Euvolämie muss bei Patienten mit kardialer Amyloidose aufrechterhalten werden. Diät- und Lebensstilmaßnahmen sind sehr wichtig. Diuretika sind der Schlüssel zur Behandlung von HF bei ATTR. Da ein übermäßiger Einsatz von Diuretika jedoch zu Hypotonie (häufig aufgrund einer autonomen Dysfunktion) führen und die klinische Situation verschlechtern kann, ist insbesondere bei ATTRm äußerste Vorsicht geboten.
Bei der Behandlung von HF bei ATTR muss berücksichtigt werden, dass eine beeinträchtigte diastolische Dysfunktion und ein verringertes Schlagvolumen zu einer kompensatorischen Tachykardie führen, um die Herzleistung aufrechtzuerhalten. Daher müssen Betablocker mit Vorsicht und individuell für jeden Patienten eingesetzt werden. Standardmäßig werden sie abgesetzt, wenn es keine Schwierigkeiten bei der Kontrolle der Herzfrequenz gibt. Dieser Ansatz ist bei ATTRwt wegen des häufigen Auftretens von Erregungsleitungsstörungen noch wichtiger.6 Kalziumantagonisten und Digoxin können sich an Amyloidfibrillen binden und sind daher bei ATTR wegen des Risikos einer Toxizität selbst in therapeutischen Dosen kontraindiziert.6
Im Gegensatz zur HF mit systolischer Dysfunktion aufgrund anderer Ätiologien gibt es keine Belege für einen prognostischen Vorteil durch den Einsatz von Betablockern, Angiotensin-Converting-Enzym-Inhibitoren oder Angiotensin-II-Rezeptor-Antagonisten bei kardialer Amyloidose. Tatsächlich kann ihr Einsatz zu einer klinischen Verschlechterung aufgrund von Hypotonie und niedrigem Output führen: In einer kürzlich erschienenen Publikation wurde über eine schlechtere Prognose bei ATTRm und eine neutrale Wirkung bei ATTRwt berichtet.34
Management von Vorhofarrhythmien
Das Management von Vorhofflimmern bei ATTR ist eine Herausforderung. Die Aufrechterhaltung eines langfristigen Sinusrhythmus ist schwierig. Eine elektrische Kardioversion kann jedoch versucht werden, da sie zu einer klinischen Verbesserung führen kann.
Das thromboembolische Risiko bei Patienten mit ATTR ist sehr hoch. Darüber hinaus kann die chronische Amyloidinfiltration zu einer mechanischen Vorhofdysfunktion führen, die bei einigen Patienten ohne Vorhofflimmern die Ursache für die Entwicklung eines Vorhofthrombus sein kann. Die Antikoagulanzientherapie bei ATTR sollte nicht auf dem CHADS2-VASC-Score basieren und sollte die Standardtherapie bei Vorhofflimmern sein. Blutungsereignisse sind seltener als bei AL, weshalb einige Krankenhäuser bei Patienten mit Sinusrhythmus eine Antikoagulanzientherapie empfehlen, wenn die Vorhoffunktion gemäß den transmissiblen Dopplergeschwindigkeiten schlecht ist. Obwohl es keine vergleichenden Studien über die Wirksamkeit direkter oraler Antikoagulanzien im Vergleich zu Vitamin-K-Antagonisten gibt, hat unser Krankenhaus ausgewählten Patienten direkte orale Antikoagulanzien verabreicht.
Rolle der Geräte
Die derzeitigen Empfehlungen für die Implantation von Herzschrittmachern sind bei ATTR und der allgemeinen Bevölkerung gleich. Einige Gruppen sprechen sich jedoch für eine prophylaktische Implantation aus, insbesondere bei Patienten mit ATTRm und Erregungsleitungsstörungen.35 Wir befürworten diese präventive Strategie nicht und haben keine so hohe Rate an Erregungsleitungsstörungen festgestellt, die eine prophylaktische Implantation bei Patienten mit ATTRm rechtfertigen würde.
Die Rolle des Einsatzes von implantierbaren Kardioverter-Defibrillatoren (ICD) bei ATTR ist nicht gut belegt. In einer kleinen Serie führte die ICD-Implantation nicht zu einer signifikanten Verbesserung der Überlebensrate, obwohl sie bei mehreren Patienten in den ersten zwei Jahren eine angemessene Wirkung hatte.36
Herztransplantation
Die Herztransplantation hat bei ATTR eine untergeordnete Rolle gespielt, da ATTRm verschiedene Organe betreffen kann und ATTRwt typischerweise ältere Patienten betrifft. Das Fehlen einer extrakardialen Beteiligung bei Patienten mit ATTRwt macht sie jedoch zu guten Kandidaten für dieses Verfahren. In der Literatur finden sich Beispiele für erfolgreiche Transplantationen bei Patienten unter 70 Jahren mit ATTRwt oder mit ATTRm und vorherrschender kardialer Beteiligung.4
Spezifische Behandlung der Transthyretin-Herzamyloidose
Derzeit gibt es keine zugelassene Therapie für die spezifische Behandlung der ATTR-Herzamyloidose, obwohl die Lebertransplantation (TxH) allein oder in Kombination mit einer Herztransplantation seit den 1990er Jahren bei ATTRm als Möglichkeit zur Beseitigung der Hauptquelle der TTR-Vorstufe eingesetzt wird.4
Lebertransplantation
Das Welttransplantationsregister für familiäre amyloidotische Polyneuropathie37 berichtet, dass sich mehr als 2000 Patienten mit ATTRm in 20 Ländern einer TxH unterzogen haben.4 Patienten mit der Val30Met-Mutation und einem überwiegend neurologischen Krankheitsbild haben eine Überlebensrate von mehr als 50 % nach 20 Jahren.3 Diese vielversprechenden Ergebnisse beruhen auf einer strengen Patientenauswahl nach Alter, Art der Mutation und Stadium der Krankheit. Die am häufigsten akzeptierte Indikation für TxH ist die Kombination aus jungem Alter, Val30Met-Mutation und frühem Krankheitsstadium.
Die wichtigsten Einschränkungen dieser Technik sind jedoch der Mangel an Spendern, die Notwendigkeit einer chronischen Immunsuppression, das fortgeschrittene Alter zum Zeitpunkt der Präsentation und die schlechteren Ergebnisse bei Patienten mit anderen Mutationen als der Val30Met-Mutation.
Außerdem wird die theoretische Unterdrückung der Produktion des mutierten Proteins durch die fortschreitende native TTR-Ablagerung nach der Implantation konterkariert,4,6 deren Mechanismus nicht vollständig verstanden ist. Tatsächlich wirkt sich die kardiale TTR-Ablagerung nach TxH auf die Morbidität und Mortalität aus.
Die Notwendigkeit, die Pathogenese von ATTR und die Grenzen von TxH besser zu verstehen, hat die Entwicklung mehrerer Medikamente angeregt.
Diese neuen Wirkstoffe setzen an verschiedenen Punkten der TTR-Amyloidogenesekaskade an (Abbildung 6). Bei der Behandlung wird es immer darum gehen, das Vorläuferprotein zu reduzieren, obwohl die Vermeidung von Ablagerungen und die Beseitigung bestehender Ablagerungen ebenso wichtig sind. Daher glauben wir, dass der Ansatz für diese Krankheit in Zukunft in Form einer kombinierten Behandlung erfolgen wird.
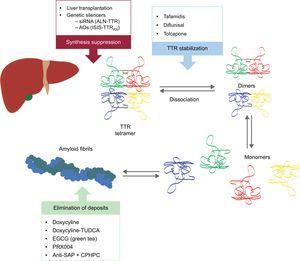
Spezifische Therapien bei kardialer Transthyretin-Amyloidose und Hauptziele. AntiSAP + CPHPC, Antiserum Amyloid-P-Komponente + (R)-1–6-oxo-hexanoyl]pyrrolidin-2-carbonsäure; AOs, Antisense-Oligonukleotide; EGCG, Epigallocatechin-3-gallat; siRNA, small interfering RNA; TTR, Transthyretin; TUDCA, Tauroursodeoxycholsäure.
Unterdrückung der Transthyretin-Synthese
Zwei Forschungslinien zur Hemmung der TTR-Expression in der Leber sind im Gange: die Verwendung von small interfering RNA (siRNA) und die Verwendung von Medikamenten mit Antisense-Oligonukleotiden (AO).
- –
Die siRNA sind doppelsträngige RNA-Moleküle, die Boten-RNA-Sequenzen zum Schweigen bringen, indem sie spezifisch an sie binden und so die Proteinbildung verhindern. Patisiran (ALN-TTR02) reduziert die TTR-Produktion um 80 %.38 Bei Patienten mit ATTRm betrug die TTR-Reduktion 87 %.39 Eine Phase-2-Studie hat vielversprechende Ergebnisse gezeigt, die stabile echokardiografische, funktionelle und analytische Parameter nach 12 und 24 Monaten belegen.40 Die Ergebnisse der neurologischen Phase-3-Studie bei Patienten mit ATTRm und einer Subanalyse von Patienten mit kardialer Beteiligung werden für 2017 erwartet (Tabelle 2). Ein weiteres Medikament, Revusiran (ALN-TTR01), wird subkutan verabreicht und unterscheidet sich von Patisiran durch die Lipid-Nanopartikel, die die siRNA verkapseln. Dieses Medikament war Gegenstand einer klinischen Studie der Phase III bei Patienten mit ATTRm, die eine Herzerkrankung haben. Die Studie wurde letztes Jahr wegen eines unerwarteten Anstiegs der Sterblichkeit in der Behandlungsgruppe abgebrochen (Tabelle 2).
Tabelle 2.Laufende klinische Studien bei Transthyretin-Herzamyloidose
Wirkmechanismus Wirkstoff Studiendesign Patienten (N) und ATTR-Subtyp Intervention Primäre Endpunkte Situation/Ergebnisse TTR-Synthesesuppression Patisiran (ALN-TTR02) NCT01961921 Phase II Studie, multizentrisch 27
ATTRm (11 Herzbeteiligungen)Patisiran 0.30 mg/kg IV alle 3 Wochen für 2 Jahre Langzeitsicherheit. Sekundäre Endpunkte: Wirkung auf neurologische Störungen und kardiale Parameter Gut verträgliches Medikament, mit ähnlichem Sicherheitsprofil im neurologischen und kardialen Phänotyp
Troponin I, NT-proBNP und echokardiographische Daten blieben nach 12 und 24 Monaten stabilNCT01960348 (APOLLO) Phase III, randomisiert, doppelblind, placebokontrolliert, multizentrisch 225
ATTRm mit neurologischer BeteiligungPatisiran Infusion IV vs. Placebo 2:1 Veränderungen im mNIS+7 Erwartet November 2017
Subanalyse von Patienten mit prognostizierter kardialer BeteiligungNCT02510261 APOLLO-Verlängerungsstudie Patisiran-Infusion IV vs. Placebo 2:1 für 52 Wochen Sicherheit und Langzeitnebenwirkungen Laufend Revusiran (ALN-TTR01) NCT02319005 (ENDEAVOUR) Phase III, randomisierte, doppelblinde, placebokontrollierte 206
ATTRm mit HerzbeteiligungRevusiran 500mg 5 d, dann wöchentlich für 2 Jahre vs. Placebo Veränderungen im 6-m-Gehtest und in den Plasma-TTR-Werten Abgebrochen wegen erhöhter Sterblichkeit in der Revusiran-Gruppe ISIS-TTRRX NCT01737398 Phase II/III, randomisierte, doppelblinde, placebokontrollierte, multizentrische 172
ATTRm mit Neuropathie; 50% gleichzeitige HerzbeteiligungISIS-TTRRX 300mg SC alle 12 h für 1 wk, dann wöchentlich für 64 Wochen vs. Placebo Veränderungen im mNIS+7 und Norfolk Fragebogen zur Lebensqualität Erwartet September 2017
Fälle von schwerer Thrombozytopenie und Blutungen berichtet
Analyse von echokardiographischen Parametern und NT-proBNP bei Patienten ohne Bluthochdruck mit LVH > 12 mmBehandlung von TTR-Kardiomyopathie mit TTR-spezifischem Antisense-Oligonukleotid Phase II, offene, nicht-randomisierte Studie 20
ATTRm mit Herzbeteiligung und ATTRwtISIS-TTRRX 300mg SC alle 12 Std./Woche Echokardiographische Parameter und kardiales MRT im Vergleich zu historischen Kontrollen Keine Verschlechterung der Belastung und verringerte LV-Masse um etwa 5%
6 Patienten schlossen 12 Monate ab; 15 Patienten 6 Monate; 1 Patient TxCNCT02627820 Phase II, open-label, nicht-randomisiert 50
ATTRwtISIS-TTRRX 300mg SC alle 12 h für 1 wk, dann 1 Woche für 18 Wochen Veränderungen der Dehnung, gemessen durch Speckle Tracking Abgebrochen ohne Beginn der Patientenrekrutierung Phase-III-Studie mit ISIS-TTRRRX zur Behandlung von TTR-Amyloid-Kardiopathie Phase III, randomisierte, doppelblinde, placebokontrollierte, multizentrische 490
ATTRwt und ATTRm mit HerzbeteiligungISIS-TTRRX 300mg SC alle 12 h für 1 Woche, dann wöchentlich für 16 Wochen mit Placebo, dann wöchentlich für 24 Monate Tod, TxC oder Einweisung wegen kardiovaskulärer Ursachen in Wartestellung Stabilisierung von TTR Tafamidis NCT01994889 Phase III, randomisierte, doppelblinde, placebokontrollierte, multizentrisch 441
ATTRwt und ATTRm mit HerzbeteiligungTafamidis 20mg oder 80mg oral alle 24 h über 30 Monate vs. Placebo All-Ursache Mortalität und kardiovaskuläre Hospitalisierung Ende Februar 2018 NCT02791230 Erweiterung Phase III NCT01994889 330
ATTRwt und ATTRm mit HerzbeteiligungTafamidis 20 mg oder 80 mg oral alle 24 h für 60 mo All-.Mortalität und Inzidenz unerwünschter Wirkungen Erwartet Dezember 2021 NCT00935012 Phase II, offene Wirksamkeits- und Sicherheitsstudie 31
ATTRwt oder ATTRm p.Val122Ile mit kardialer BeteiligungTafamidis 20mg oral Sicherheit und Wirksamkeit Laufend bis Dezember 2021 Diflunisal NCT00294671 Phase III, randomisiert, doppelblind, Placebo-kontrolliert, multizentrisch 130
ATTRm mit neurologischem Phänotyp (50% mit kardialer Beteiligung)Diflunisal 250mg oral alle 12 h vs. Placebo über 24 Monate NIS+7 nach 24 Monaten NIS+7 Diflunisal vs. Placebo 16.3 (P Keine Verringerung der ventrikulären Dicke oder Belastung bei Patienten mit Herzbeteiligung gegenüber Placebo Elimination von Ablagerungen Doxycyclin + TUDCA/UDCA NCT01171859 Phase II, Open-Label, nicht-randomisiert, prospektiv 40
ATTR (25 ATTRm, 13 ATTRwt und 2 Domino-Lebertransplantat-Empfänger)Doxycyclin 100 mg alle 12 h + TUDCA 250 mg alle 8 h für 12 mo, dann 6 Monate ohne Therapie Verbesserung 14 Patienten zogen sich zurück
Nebenwirkungen der Haut, 16 Patienten
68% der 25 auswertbaren Patienten erfüllten den primären Endpunkt
Gesamtverbesserung der Belastung nach 12 Monaten und Verschlechterung nach 6 Monaten ohne TherapieNCT01855360 Phase II, offene, nicht-randomisierte, prospektive Studie im Vergleich zu historischen Kontrollen 30
Herz-ATTR-Amyloidose (27 ATTRwt und 3 ATTRm). Historische Kontrollen, 14 ATTRwt-PatientenDoxycyclin 100 mg alle 12 Stunden + TUDCA 250 mg alle 8 Stunden für 18 Monate Veränderungen der Längsdehnung alle 6 Monate 22 Patienten schlossen die Studie ab und waren auswertbar
Größere Verschlechterung der Dehnung in den Kontrollen gegenüber der Behandlungsgruppe
Erhöhter NT-proBNP in der Behandlungsgruppe; bei Kontrollen nicht gemessenNCT01677286 Phase II, offen, nicht randomisiert, prospektiv 25
Systemische Amyloidose (6 ATTRwt und 3 ATTRm)Doxycyclin 100 mg alle 12 h für 12 Monate Medikamentensicherheit
Ansprechen der betroffenen OrganeVerschlechterung von NT-proBNP und Nierenfunktion
Keine Verbesserung bei anderen untersuchten Parametern
60% der Patienten hatten Hautkomplikationen und 30% zogen sich aufgrund von Haut- oder Magen-Darm-Problemen zurückNCT01171859 Phase II, offene, nicht-randomisierte, prospektive 45
35 mit Herzbeteiligung; 25 ATTRm; 5 ATTRm mit TxH; 13 ATTRwt; und 2 Domino-Lebertransplantat-EmpfängerDoxycyclin 100 mg alle 12 Stunden + TUDCA 250 mg alle 8 Stunden für 12 Monate
Anschließende Nachbeobachtungsphase ohne Behandlung für 6 MonateAnsprechen auf die Behandlung definiert als Kardiales Ansprechen bei 25 Patienten
68% hatten kardiales Ansprechen
Erhöhte NT-proBNP und Verschlechterung der Belastung während der Nachbeobachtung ohne Behandlung
Hohe Anzahl von Abbrüchen aufgrund von unerwünschten Wirkungen
14 Abbrüche in der Behandlungsphase und 5 Abbrüche in der Phase ohne BehandlungWirkung von Doxycyclin + UDCA auf ATTR Phase II, open-label, nicht-randomisiert, prospektiv 28
ATTR mit kardialer Beteiligung (27 ATTRm und 1 ATTRwt)Doxycyclin 200 mg/d für 4 Wochen, dann 2 Wochen ausgesetzt, dann UDCA 750 mg/d für 12 Monate
Anschließende Nachbeobachtungsphase ohne Behandlung für 6 MonateVeränderungen im NT-proBNP und Kumamoto-Score Nur 14% schlossen die Studie ab und 36% schlossen 12 Monate ab
Keine Veränderungen im NT-proBNP nach 6 Monaten und Verschlechterung nach 12 Monaten
Stabile LVH
Verschlechterung des Kumamoto-Scores nach 12 MonatenEGCG NCT01171859 Phase II, Open-Label, nicht-randomisiert, prospektiv 25
ATTRwt600 mg, EGCG für 12 Monate Veränderungen im ECHO und Herz-MRT (n = 14) Rückläufige LV-Masse 6% durch Herz-MRT (P = 0.03)
LVEF, Myokarddicke und MAPSE im ECHO unverändertAntiSAP + CPHPC NCT03044353 Phase II, open-label, randomisiert 40
Kohorte 1: kardiale ATTR-Amyloidose
Kohorte 2: Primäre Amyloidose nach 6 Monaten ChemotherapieAnti-SAP + CPHPC monatlich für 6 Monate Reduzierte Amyloidlast durch kardiales MRT und ECHO Start in 2017 AntiSAP + CPHPC, Antiserum-Amyloid-P-Komponente + (R)-1–6-oxo-hexanoyl]pyrrolidin-2-carbonsäure; ATTRm, mutierte Transthyretin-Amyloidose; ATTRwt, Wildtyp-Transthyretin-Amyloidose; BNP, natriuretisches Hirnpeptid; ECHO, Echokardiogramm; EGCG, Epigallocatechin-3-Gallat; IV, intravenös; LV, linker Ventrikel; LVEF, linksventrikuläre Auswurffraktion; LVH, linksventrikuläre Hypertrophie; MAPSE, systolische Exkursion der mitralen Anularebene; mNIS, Modified Neuropathy Impairment Score; MRI, Magnetresonanztomographie; NIS, Neuropathy Impairment Score; NIS-LL, Neuropathy Impairment Score of the Lower Limbs; NT-proBNP, amino-terminales pro-brain natriuretisches Peptid; SC, subkutan; TTR, Transthyretin; TUDCA, Tauroursodeoxycholsäure; TxC, Herztransplantation; TxH, Lebertransplantation; UDCA, Ursodeoxycholsäure.
- –
Die AOs sind kurze Oligonukleotidstränge, die spezifisch an RNA binden und so die Translation und die Zielproteinsynthese verhindern.4 ISIS-TTRRX ist ein subkutan zu verabreichendes AO, das bei gesunden Probanden nachweislich zu einer dosisabhängigen Senkung der TTR-Werte um 75 % bis 90 % führt.4 Die Phase-III-Studie bei Patienten mit ATTRm und neurologischem Phänotyp endete im März 2017, und ihre Ergebnisse werden bis Ende 2017 erwartet. Die US-amerikanische Arzneimittelbehörde FDA verschob jedoch den Beginn einer Phase-III-Studie bei Patienten mit ATTRwt und ATTRm mit Herzerkrankungen aufgrund von Fällen schwerer Thrombozytopenie in der neurologischen Studie (Tabelle 2). Da 50 % der Teilnehmer an der neurologischen Studie eine Herzerkrankung hatten, werden die Ergebnisse dieser kardiologischen Teilstudie darüber entscheiden, ob die Phase-III-Studie wieder aufgenommen wird. Andererseits gibt es vorläufige Daten aus einer offenen Phase-II-Studie. In dieser Studie erhielten 22 Patienten mit ATTRwt und ATTRm mit Herzerkrankungen eine wöchentliche Injektion von ISIS-TTRRX. Dem Bericht zufolge ist das Sicherheitsprofil des Medikaments sehr günstig, und die Zwischendaten zum Fortschreiten der Herzerkrankung durch CMR, NT-proBNP und 6-Minuten-Tests sind positiv.41
Stabilisierung von Transthyretin
Die Aufspaltung des TTR-Tetramers in Untereinheiten ist ein entscheidender Schritt bei der ATTR-Fibrillenbildung. Diflunisal und Tafamidis sind zwei TTR-Stabilisatoren mit nachgewiesener Wirksamkeit bei ATTRm-Polyneuropathie.
- –
Tafamidis ist ein oral verabreichtes kleines Molekül, das an TTR an T4-Bindungsstellen bindet, indem es das Protein stabilisiert und seine Dissoziation verhindert. Nach der Veröffentlichung der Ergebnisse einer randomisierten Doppelblindstudie an 125 Patienten mit ATTRm und der Val30Met-Mutation im Anfangsstadium der neurologischen Erkrankung42 genehmigte die Europäische Arzneimittel-Agentur 2011 seine Verwendung als Arzneimittel für seltene Leiden zur Verzögerung der neurologischen Progression. Jüngste Daten belegen die Wirksamkeit des Medikaments im Hinblick auf eine neurologische Stabilität bei mindestens 60 % der Teilnehmer nach einer Nachbeobachtungszeit von mehr als vier Jahren. Bislang wurde es nur in begrenztem Umfang bei ATTR und kardiologischen Erkrankungen eingesetzt. Eine Phase-II-Studie an 21 Patienten mit ATTRm und verschiedenen Mutationen zeigte, dass NT-proBNP und echokardiografische Parameter nach 12 Monaten stabil blieben.43 Daten aus einer 5-Jahres-Kohortenstudie bestätigten, dass das Medikament in einer Dosis von 20 mg gut verträglich war, obwohl nur wenige Patienten mit ATTRwt nach 3,5 Jahren stabil blieben.44 Die ATTR-ACT-Studie ist eine 30-monatige Phase-III-Studie, in der die Wirksamkeit, Sicherheit und Verträglichkeit von Tafamidis in einer Dosierung von 20 mg und 80 mg im Vergleich zu Placebo bei 440 Patienten mit ATTRm, ATTRwt und HF untersucht wird. Der primäre Endpunkt umfasst die Krankenhaussterblichkeit und -aufnahme. Die Ergebnisse werden 2018 erwartet.3,27
- –
Diflunisal ist ein nichtsteroidales entzündungshemmendes Mittel, das TTR-Moleküle in vitro stabilisiert. Es ist in Spanien nicht erhältlich, kann aber aus dem Ausland als „compassionate use“ angefordert werden. Eine Phase-III-Studie zu ATTRm bei Patienten mit überwiegend neurologischer Beteiligung, von denen mehr als die Hälfte eine Herzerkrankung hatte, ergab keine signifikanten Unterschiede bei den echokardiografischen Parametern im Studienzeitraum (Tabelle 2).45 Aufgrund seines Potenzials für gastrointestinale Nebenwirkungen, Nierenversagen, Wasserretention und Bluthochdruck ist es für Patienten mit Herzerkrankungen nicht geeignet. Die Evidenz zu Diflunisal bei Patienten mit ATTR ist sehr begrenzt. Es gibt eine Studie, die jedoch dadurch eingeschränkt ist, dass es sich um eine nicht-randomisierte Studie eines einzigen Zentrums mit geringer Nachbeobachtung und wenigen Patienten (n = 13) handelt. Es gab keine Einweisungen wegen dekompensierter HF, aber es kam zu einer signifikanten Verschlechterung der Nierenfunktion.46
- –
Kürzlich wies eine spanische Gruppe nach, dass Tolcapon (ein oraler Katechol-O-Methyltransferase-Hemmer, der bei der Behandlung der Parkinson-Krankheit eingesetzt wird) die Fähigkeit besitzt, in vitro an das TTR von Patienten mit ATTRwt und Val122Ile mit höherer Affinität als andere Stabilisatoren zu binden.47
Elimination von Amyloid-Ablagerungen
Amyloid-Ablagerungen sind sehr stabil und es scheint, dass der menschliche Organismus kaum in der Lage ist, sie zu beseitigen. Behandlungen, die die Produktion neuer Amyloide verhindern, wie die Chemotherapie bei AL, können die Ablagerungen jedoch allmählich und mit unterschiedlichen organspezifischen Raten abbauen. Die kardiale Clearance ist besonders gering, und bisher gibt es kaum Hinweise auf eine Rückbildung. Mehrere Moleküle werden derzeit untersucht, um die kardiale Amyloid-Clearance bei ATTR zu beschleunigen:
- –
Doxycyclin (ein häufig verwendetes Antibiotikum) unterbricht die Bildung von Amyloidfibrillen. Die synergistische Wirkung der Kombination von Doxycyclin und Tauroursodeoxychol-Gallensäure (TUDCA), die bei der Behandlung von Lebererkrankungen eingesetzt wird, hat in Tiermodellen gezeigt, dass sie TTR-Ablagerungen beseitigt. Eine Phase-II-Studie mit 20 Patienten zeigte nach einjähriger Behandlung mit Doxycyclin/TUDCA keine kardiale oder neurologische Progression bei akzeptablem Sicherheits- und Verträglichkeitsprofil.4 In anderen Phase-II-Studien wurde versucht, diese Ergebnisse mit der Kombination Doxycyclin/TUDCA, Doxycyclin/Ursodeoxycholsäure oder Doxycyclin allein zu bestätigen.48-50 Die vorläufigen Ergebnisse einer dieser Studien deuten auf eine schützende Wirkung hin, mit einer geringeren Verschlechterung der Herzfunktion aufgrund der Belastung in der Behandlungsgruppe. Eine andere dieser Studien kam bei 40 Patienten mit ATTR zu ähnlichen Ergebnissen: NT-proBNP, Funktionsklasse, LVEF und Myokarddicke waren nach 12 Monaten stabil (Tabelle 2). Alle diese Studien wiesen jedoch eine hohe Abbruchrate auf (35%-44%), hauptsächlich aufgrund von unerwünschten Wirkungen, insbesondere Sonnenüberempfindlichkeit und Magen-Darm-Beschwerden (bis zu 30%).48-50
- –
Das EGCG (Epigallocatechin-3-Gallat) ist das am häufigsten vorkommende Katechin in grünem Tee und hat in vitro und in einem Mausmodell gezeigt, dass es die Amyloidbildung hemmt und bestehende Ablagerungen beseitigt.4 Das CMRI zeigte, dass die tägliche Verabreichung von 600 mg EGCG in einer Gruppe von 25 Patienten mit einer Stabilisierung der linksventrikulären Masse verbunden war (Tabelle 2).51
- –
Der PRX004 ist ein monoklonaler Antikörper, der durch Bindung an monomerspezifische Epitope und fehlgefaltetes TTR wirkt. Auf diese Weise löst er die Beseitigung von Ablagerungen durch Aktivierung der Phagozytose aus.52 Die Grundlage seines Wirkmechanismus ähnelt dem eines bei AL verwendeten Antikörpers. Phase-II-Studien mit diesem Antikörper zeigen vielversprechende Ergebnisse. Eine Phase-I-II-Studie mit diesem neuen Antikörper soll 2017 beginnen.
- –
Unabhängig vom Typ des Amyloid-Vorläuferproteins enthalten alle Ablagerungen die Serum-Amyloid-Komponente P (SAP). Unter Verwendung dieses Moleküls als Zielmolekül wurde gezeigt, dass Anti-SAP-Antikörper eine Makrophagen-vermittelte und Komplement-abhängige Reaktion auslösen, die in einem Mausmodell zu einer weitgehenden Beseitigung viszeraler Amyloidablagerungen führte. Die Bis-D-Prolin-Verbindung CPHPC kann Plasma-SAP neutralisieren, und die gleichzeitige Verabreichung mit Anti-SAP-IgG ermöglicht es dem Antikörper, SAP-haltige Ablagerungen im Gewebe zu erreichen.53 In einer 2015 veröffentlichten Phase-I-Studie wurde bei 15 Patienten mit systemischer Amyloidose ohne Herzbeteiligung die Beseitigung hepatischer Ablagerungen mit nur wenigen unerwünschten Wirkungen nachgewiesen.53 Eine Phase-II-Studie mit Patienten mit ATTR-Herzamyloidose und AL soll 2017 beginnen (Tabelle 2).
ZUSAMMENFASSUNGEN
Transthyretin-Herzamyloidose wird immer häufiger diagnostiziert. 99mTc-DPD-Szintigraphie und CMRI sind Beispiele für Techniken, die zur einfachen und frühzeitigen Identifizierung von Patienten mit ATTR eingesetzt werden können.
Mehrere ATTR-spezifische Medikamente befinden sich derzeit in der Endphase der Forschung. Daher glauben wir, dass die ATTR-Herzamyloidose bald als behandelbare Entität und nicht als tödliche Krankheit angesehen werden wird.
Finanzierung
Diese Arbeit wurde mit teilweiser Unterstützung des Carlos III Health Institute und der Spanischen Gesellschaft für Kardiologie (Forschungsstipendium 2016 für E. González-López) durchgeführt. Die Unterstützung des Carlos III Health Institute wird durch den Europäischen Fonds für regionale Entwicklung „Another Way to Make Europe“ finanziert.
CONFLICTS OF INTEREST
E. González-López hat als Referent an von Pfizer organisierten Veranstaltungen teilgenommen. P. Garcia-Pavia erhielt Zahlungen als Redner bei von Pfizer organisierten Veranstaltungen und als Berater von Alnylam, Prothena und Pfizer. E. González-López, A. López-Sainz und P. Garcia-Pavia erklären, dass Pfizer Forschungsprojekte ihrer Einrichtung finanziert hat.